

Case report: Extending the spectrum of clinical and molecular findings in FOXC1 haploinsufficiency syndrome
- PMID: 37424725
- PMCID: PMC10326848
- DOI: 10.3389/fgene.2023.1174046
Case report: Extending the spectrum of clinical and molecular findings in FOXC1 haploinsufficiency syndrome
Abstract
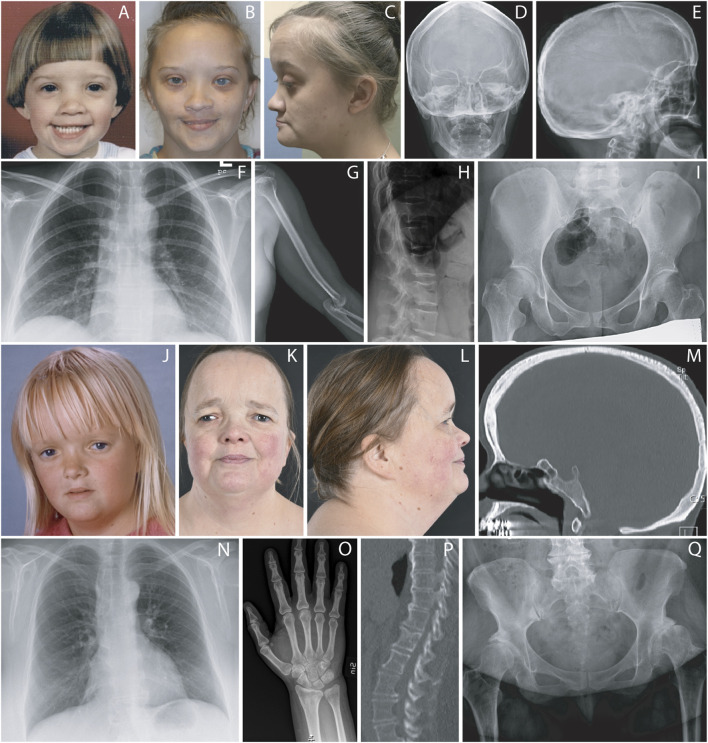
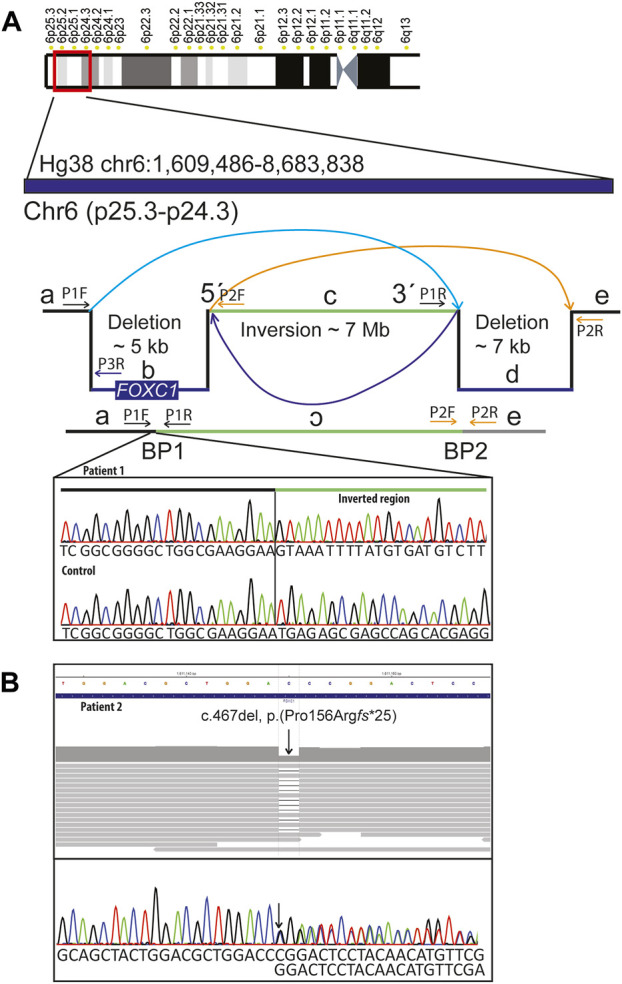
FOXC1 is a ubiquitously expressed forkhead transcription factor that plays a critical role during early development. Germline pathogenic variants in FOXC1 are associated with anterior segment dysgenesis and Axenfeld-Rieger syndrome (ARS, #602482), an autosomal dominant condition with ophthalmologic anterior segment abnormalities, high risk for glaucoma and extraocular findings including distinctive facial features, as well as dental, skeletal, audiologic, and cardiac anomalies. De Hauwere syndrome is an ultrarare condition previously associated with 6p microdeletions and characterized by anterior segment dysgenesis, joint instability, short stature, hydrocephalus, and skeletal abnormalities. Here, we report clinical findings of two unrelated adult females with FOXC1 haploinsufficiency who have ARS and skeletal abnormalities. Final molecular diagnoses of both patients were achieved using genome sequencing. Patient 1 had a complex rearrangement involving a 4.9 kB deletion including FOXC1 coding region (Hg19; chr6:1,609,721-1,614,709), as well as a 7 MB inversion (Hg19; chr6:1,614,710-8,676,899) and a second deletion of 7.1 kb (Hg19; chr6:8,676,900-8,684,071). Patient 2 had a heterozygous single nucleotide deletion, resulting in a frameshift and a premature stop codon in FOXC1 (NM_001453.3): c.467del, p.(Pro156Argfs*25). Both individuals had moderate short stature, skeletal abnormalities, anterior segment dysgenesis, glaucoma, joint laxity, pes planovalgus, dental anomalies, hydrocephalus, distinctive facial features, and normal intelligence. Skeletal surveys revealed dolichospondyly, epiphyseal hypoplasia of femoral and humeral heads, dolichocephaly with frontal bossin gand gracile long bones. We conclude that haploinsufficiency of FOXC1 causes ARS and a broad spectrum of symptoms with variable expressivity that at its most severe end also includes a phenotype overlapping with De Hauwere syndrome.
Keywords: Axenfeld-Rieger Syndrome; De Hauwere Syndrome; FOXC1; case report; genome sequencing; skeletal anomalies.
Copyright © 2023 Garza Flores, Nordgren, Pettersson, Dias-Santagata, Nilsson, Hammarsjö, Lindstrand, Batkovskyte, Wiggs, Walton, Goldenberg, Eisfeldt, Lin, Lachman, Nishimura and Grigelioniene.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
PITX2 and FOXC1 spectrum of mutations in ocular syndromes.Eur J Hum Genet. 2012 Dec;20(12):1224-33. doi: 10.1038/ejhg.2012.80. Epub 2012 May 9. Eur J Hum Genet. 2012. PMID: 22569110 Free PMC article.
-
Cardiac anomalies in Axenfeld-Rieger syndrome due to a novel FOXC1 mutation.Am J Med Genet A. 2013 Jan;161A(1):114-9. doi: 10.1002/ajmg.a.35697. Epub 2012 Dec 14. Am J Med Genet A. 2013. PMID: 23239455
-
Axenfeld-Rieger syndrome: more than meets the eye.J Med Genet. 2023 Apr;60(4):368-379. doi: 10.1136/jmg-2022-108646. Epub 2022 Jul 26. J Med Genet. 2023. PMID: 35882526 Free PMC article.
-
Ring chromosome 6 in a child with anterior segment dysgenesis and review of its overlap with other FOXC1 deletion phenotypes.Congenit Anom (Kyoto). 2019 Sep;59(5):174-178. doi: 10.1111/cga.12309. Epub 2018 Oct 9. Congenit Anom (Kyoto). 2019. PMID: 30225942 Review.
-
The 6p25 deletion syndrome: An update on a rare neurocristopathy.Ophthalmic Genet. 2017 Mar-Apr;38(2):101-107. doi: 10.3109/13816810.2016.1164191. Epub 2016 Apr 12. Ophthalmic Genet. 2017. PMID: 27070436 Review.
Cited by
-
Congenital anterior segment ocular disorders: Genotype-phenotype correlations and emerging novel mechanisms.Prog Retin Eye Res. 2024 Sep;102:101288. doi: 10.1016/j.preteyeres.2024.101288. Epub 2024 Aug 2. Prog Retin Eye Res. 2024. PMID: 39097141 Review.
-
Alternative Genetic Diagnoses in Axenfeld-Rieger Syndrome Spectrum.Genes (Basel). 2023 Oct 17;14(10):1948. doi: 10.3390/genes14101948. Genes (Basel). 2023. PMID: 37895297 Free PMC article.
-
FOXC1 and FOXC2 regulate growth plate chondrocyte maturation towards hypertrophy in the embryonic mouse limb skeleton.Development. 2024 Aug 15;151(16):dev202798. doi: 10.1242/dev.202798. Epub 2024 Aug 22. Development. 2024. PMID: 39012257 Free PMC article.
-
Diagnostic Challenges of Axenfeld-Rieger Syndrome and a Novel FOXC1 Gene Mutation in a Polish Family.J Clin Med. 2024 Sep 27;13(19):5761. doi: 10.3390/jcm13195761. J Clin Med. 2024. PMID: 39407821 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous