

This is a preprint.
Extensible benchmarking of methods that identify and quantify polyadenylation sites from RNA-seq data
- PMID: 37425672
- PMCID: PMC10327023
- DOI: 10.1101/2023.06.23.546284
Extensible benchmarking of methods that identify and quantify polyadenylation sites from RNA-seq data
Update in
-
Extensible benchmarking of methods that identify and quantify polyadenylation sites from RNA-seq data.RNA. 2023 Dec;29(12):1839-1855. doi: 10.1261/rna.079849.123. Epub 2023 Oct 10. RNA. 2023. PMID: 37816550 Free PMC article. Review.
Abstract
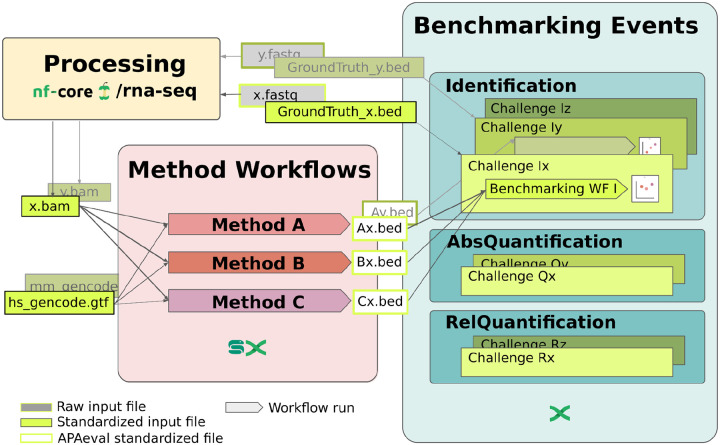
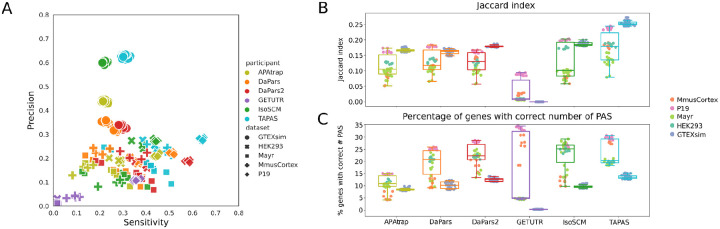
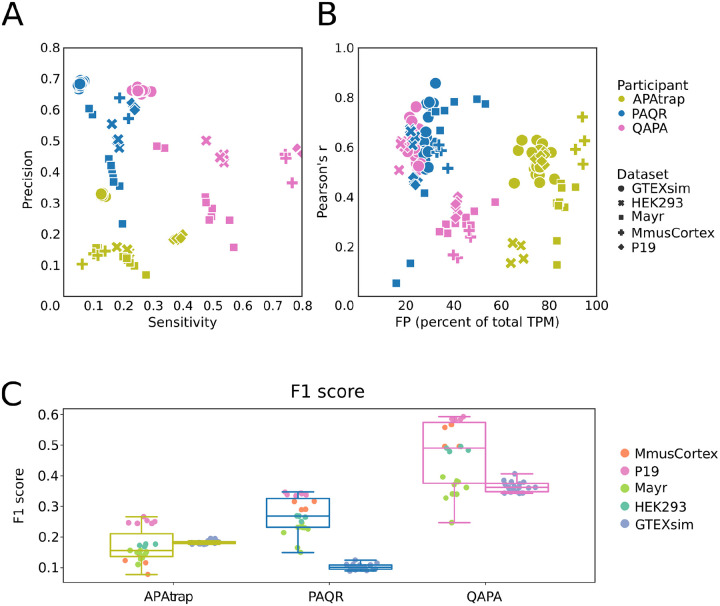
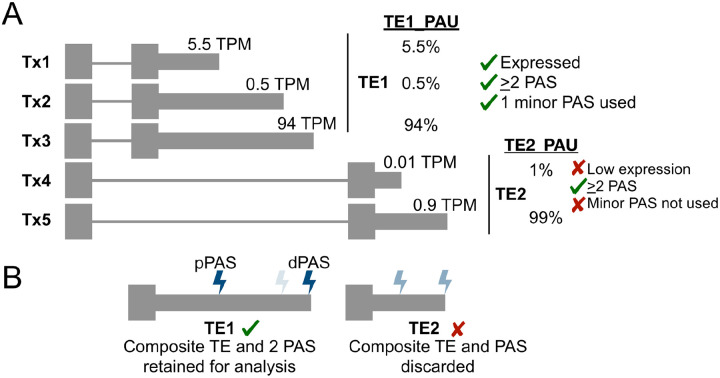
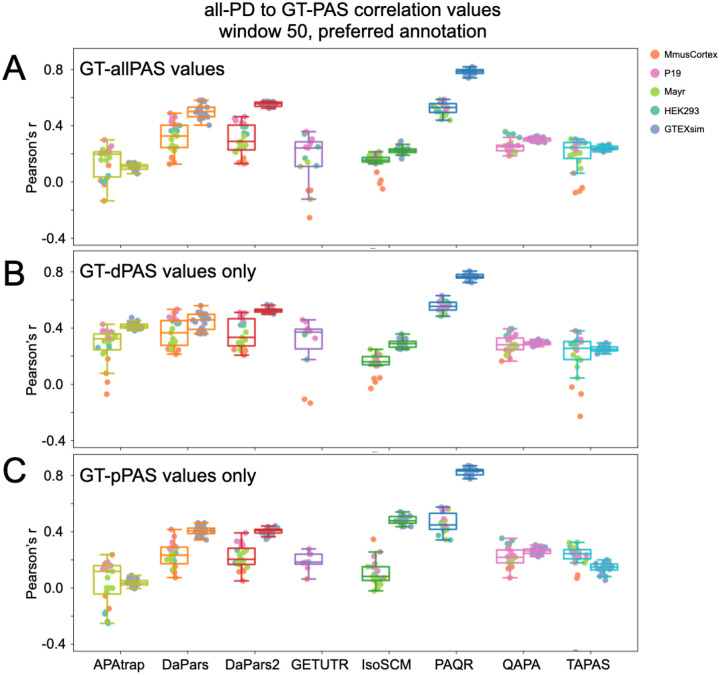
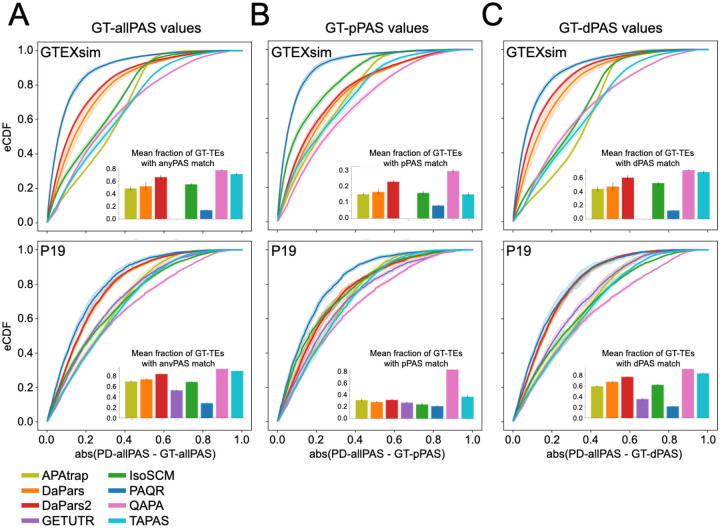
The tremendous rate with which data is generated and analysis methods emerge makes it increasingly difficult to keep track of their domain of applicability, assumptions, and limitations and consequently, of the efficacy and precision with which they solve specific tasks. Therefore, there is an increasing need for benchmarks, and for the provision of infrastructure for continuous method evaluation. APAeval is an international community effort, organized by the RNA Society in 2021, to benchmark tools for the identification and quantification of the usage of alternative polyadenylation (APA) sites from short-read, bulk RNA-sequencing (RNA-seq) data. Here, we reviewed 17 tools and benchmarked eight on their ability to perform APA identification and quantification, using a comprehensive set of RNA-seq experiments comprising real, synthetic, and matched 3'-end sequencing data. To support continuous benchmarking, we have incorporated the results into the OpenEBench online platform, which allows for seamless extension of the set of methods, metrics, and challenges. We envisage that our analyses will assist researchers in selecting the appropriate tools for their studies. Furthermore, the containers and reproducible workflows generated in the course of this project can be seamlessly deployed and extended in the future to evaluate new methods or datasets.
Keywords: (alternative) polyadenylation; Benchmarking; RNA-seq; bioinformatics; community initiative.
Figures

References
-
- Capella-Gutierrez S., de la Iglesia D., Haas J., Lourenco A., Fernández J. M., Repchevsky D., Dessimoz C., Schwede T., Notredame C., Gelpi J. L., & Valencia A. (2017). Lessons Learned: Recommendations for Establishing Critical Periodic Scientific Benchmarking (p. 181677). bioRxiv. 10.1101/181677 - DOI
-
- Da Veiga Leprevost F., Grüning B. A., Alves Aflitos S., Röst H. L., Uszkoreit J., Barsnes H., Vaudel M., Moreno P., Gatto L., Weber J., Bai M., Jimenez R. C., Sachsenberg T., Pfeuffer J., Vera Alvarez R., Griss J., Nesvizhskii A. I., & Perez-Riverol Y. (2017). BioContainers: An open-source and community-driven framework for software standardization. Bioinformatics, 33(16), 2580–2582. 10.1093/bioinformatics/btx192 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources