

This is a preprint.
CHARR efficiently estimates contamination from DNA sequencing data
- PMID: 37425834
- PMCID: PMC10327099
- DOI: 10.1101/2023.06.28.545801
CHARR efficiently estimates contamination from DNA sequencing data
Update in
-
CHARR efficiently estimates contamination from DNA sequencing data.Am J Hum Genet. 2023 Dec 7;110(12):2068-2076. doi: 10.1016/j.ajhg.2023.10.011. Epub 2023 Nov 23. Am J Hum Genet. 2023. PMID: 38000370 Free PMC article.
Abstract
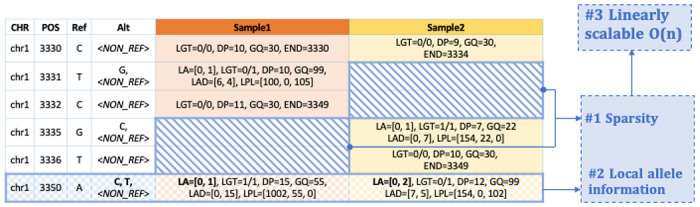
DNA sample contamination is a major issue in clinical and research applications of whole genome and exome sequencing. Even modest levels of contamination can substantially affect the overall quality of variant calls and lead to widespread genotyping errors. Currently, popular tools for estimating the contamination level use short-read data (BAM/CRAM files), which are expensive to store and manipulate and often not retained or shared widely. We propose a new metric to estimate DNA sample contamination from variant-level whole genome and exome sequence data, CHARR, Contamination from Homozygous Alternate Reference Reads, which leverages the infiltration of reference reads within homozygous alternate variant calls. CHARR uses a small proportion of variant-level genotype information and thus can be computed from single-sample gVCFs or callsets in VCF or BCF formats, as well as efficiently stored variant calls in Hail VDS format. Our results demonstrate that CHARR accurately recapitulates results from existing tools with substantially reduced costs, improving the accuracy and efficiency of downstream analyses of ultra-large whole genome and exome sequencing datasets.
Figures






References
-
- Bergström A., McCarthy S. A., Hui R., Almarri M. A., Ayub Q., Danecek R., Chen Y, Felkel S., Hallast R., Kamm J., Blanché H., Deleuze J.-F., Cann H., Mallick S., Reich D., Sandhu M. S., Skoglund P., Scally A., Xue Y, … Tyler-Smith C. (2020). Insights into human genetic variation and population history from 929 diverse genomes. Science, 367(6484). 10.1126/science.aay5012 - DOI - PMC - PubMed
-
- Chen S., Francioli L. C., Goodrich J. K., Collins R. L., Kanai M., Wang Q., Alföldi J., Watts N. A., Vittal C., Gauthier L. D., Poterba T., Wilson M. W., Tarasova Y, Phu W., Yohannes M. T., Koenig Z., Farjoun Y, Banks E., Donnelly S., … Karczewski K. J.. (2022). A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. In bioRxiv (p. 2022.03.20.485034). 10.1101/2022.03.20.485034 - DOI
-
- Hail Team. (2023). Hail 0.2.106-a6c75d687a19. https://github.com/hail-is/hail/commit/a6c75d687a19
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous