

This is a preprint.
The importance of input sequence set to consensus-derived proteins and their relationship to reconstructed ancestral proteins
- PMID: 37425932
- PMCID: PMC10327145
- DOI: 10.1101/2023.06.29.547063
The importance of input sequence set to consensus-derived proteins and their relationship to reconstructed ancestral proteins
Update in
-
The importance of input sequence set to consensus-derived proteins and their relationship to reconstructed ancestral proteins.Protein Sci. 2024 Jun;33(6):e5011. doi: 10.1002/pro.5011. Protein Sci. 2024. PMID: 38747388 Free PMC article.
Abstract
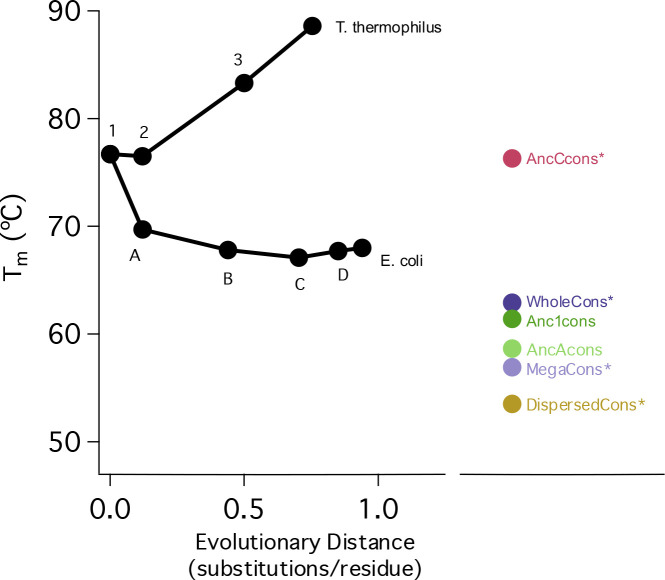
A protein sequence encodes its energy landscape - all the accessible conformations, energetics, and dynamics. The evolutionary relationship between sequence and landscape can be probed phylogenetically by compiling a multiple sequence alignment of homologous sequences and generating common ancestors via Ancestral Sequence Reconstruction or a consensus protein containing the most common amino acid at each position. Both ancestral and consensus proteins are often more stable than their extant homologs - questioning the differences and suggesting that both approaches serve as general methods to engineer thermostability. We used the Ribonuclease H family to compare these approaches and evaluate how the evolutionary relationship of the input sequences affects the properties of the resulting consensus protein. While the overall consensus protein is structured and active, it neither shows properties of a well-folded protein nor has enhanced stability. In contrast, the consensus protein derived from a phylogenetically-restricted region is significantly more stable and cooperatively folded, suggesting that cooperativity may be encoded by different mechanisms in separate clades and lost when too many diverse clades are combined to generate a consensus protein. To explore this, we compared pairwise covariance scores using a Potts formalism as well as higher-order couplings using singular value decomposition (SVD). We find the SVD coordinates of a stable consensus sequence are close to coordinates of the analogous ancestor sequence and its descendants, whereas the unstable consensus sequences are outliers in SVD space.
Keywords: Ancestral Sequence Reconstruction; Consensus Design; Protein Folding; Protein Stability; Singular Value Decomposition.
Figures









References
-
- Thornton JW, Need E, Crews D (2003) Resurrecting the ancestral steroid receptor: Ancient origin of estrogen signaling. Science (80-. ). 301:1714–1717. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources