

This is a preprint.
Fast and flexible joint fine-mapping of multiple traits via the Sum of Single Effects model
- PMID: 37425935
- PMCID: PMC10327118
- DOI: 10.1101/2023.04.14.536893
Fast and flexible joint fine-mapping of multiple traits via the Sum of Single Effects model
Abstract
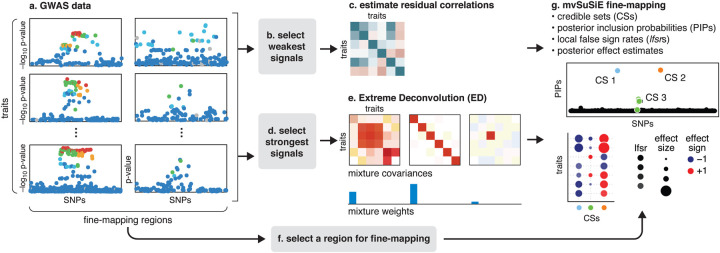
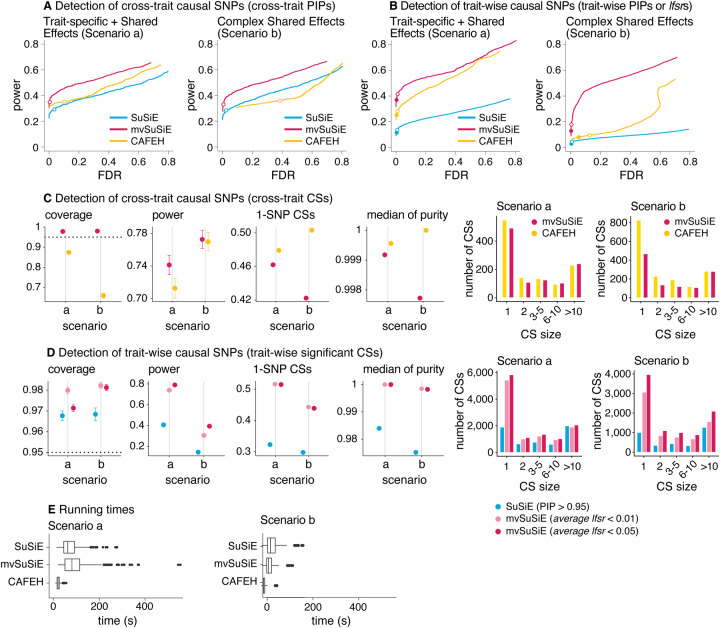
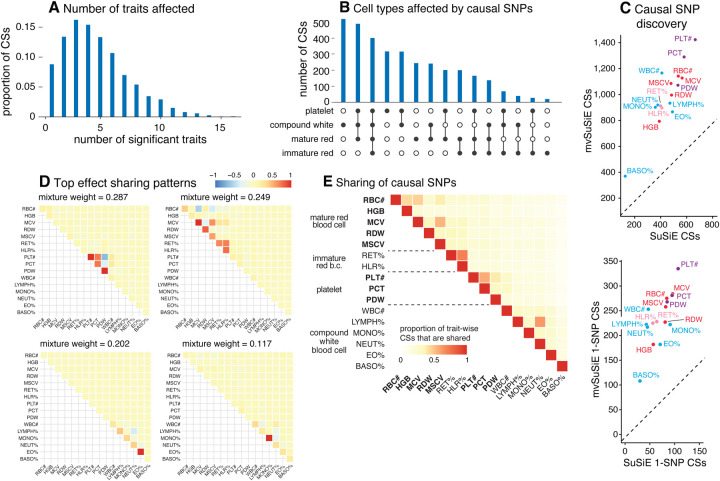
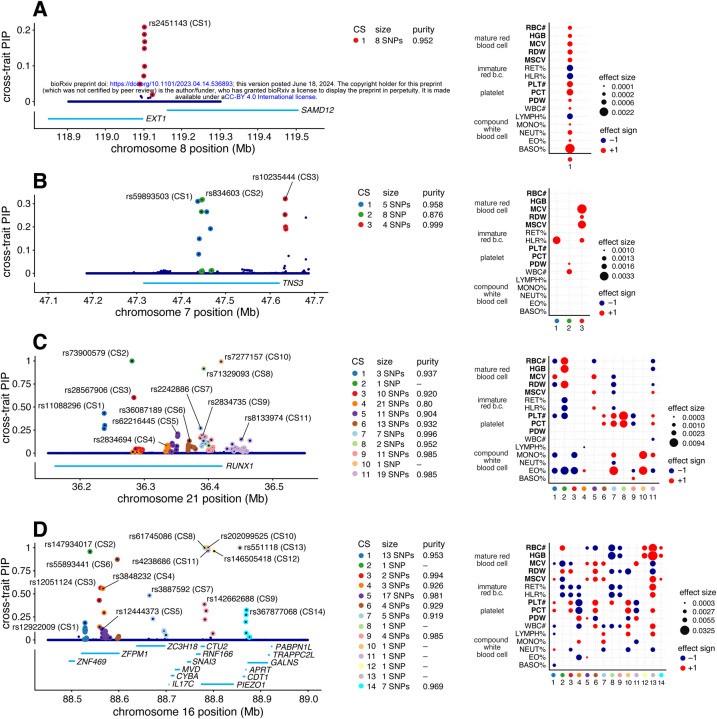
We introduce mvSuSiE, a multi-trait fine-mapping method for identifying putative causal variants from genetic association data (individual-level or summary data). mvSuSiE learns patterns of shared genetic effects from data, and exploits these patterns to improve power to identify causal SNPs. Comparisons on simulated data show that mvSuSiE is competitive in speed, power and precision with existing multi-trait methods, and uniformly improves on single-trait fine-mapping (SuSiE) in each trait separately. We applied mvSuSiE to jointly fine-map 16 blood cell traits using data from the UK Biobank. By jointly analyzing the traits and modeling heterogeneous effect sharing patterns, we discovered a much larger number of causal SNPs (>3,000) compared with single-trait fine-mapping, and with narrower credible sets. mvSuSiE also more comprehensively characterized the ways in which the genetic variants affect one or more blood cell traits; 68% of causal SNPs showed significant effects in more than one blood cell type.
Conflict of interest statement
Competing interests. The authors declare no competing financial interests.
Figures
Similar articles
-
Multivariate Bayesian variable selection for multi-trait genetic fine mapping.J R Stat Soc Ser C Appl Stat. 2024 Oct 28;74(2):331-351. doi: 10.1093/jrsssc/qlae055. eCollection 2025 Mar. J R Stat Soc Ser C Appl Stat. 2024. PMID: 40092670
-
BEATRICE: Bayesian fine-mapping from summary data using deep variational inference.Bioinformatics. 2024 Oct 1;40(10):btae590. doi: 10.1093/bioinformatics/btae590. Bioinformatics. 2024. PMID: 39360993 Free PMC article.
-
BAYESIAN VARIABLE SELECTION IN A COX PROPORTIONAL HAZARDS MODEL WITH THE "SUM OF SINGLE EFFECTS" PRIOR.ArXiv [Preprint]. 2025 Jun 6:arXiv:2506.06233v1. ArXiv. 2025. PMID: 40503016 Free PMC article. Preprint.
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
-
Management of faecal incontinence and constipation in adults with central neurological diseases.Cochrane Database Syst Rev. 2013 Dec 18;(12):CD002115. doi: 10.1002/14651858.CD002115.pub4. Cochrane Database Syst Rev. 2013. Update in: Cochrane Database Syst Rev. 2014 Jan 13;(1):CD002115. doi: 10.1002/14651858.CD002115.pub5. PMID: 24347087 Updated.
Cited by
-
Integration of expression QTLs with fine mapping via SuSiE.PLoS Genet. 2024 Jan 25;20(1):e1010929. doi: 10.1371/journal.pgen.1010929. eCollection 2024 Jan. PLoS Genet. 2024. PMID: 38271473 Free PMC article.
References
-
- Tam V., Patel N., Turcotte M., Bossé Y., Paré G. and Meyre D., “Benefits and limitations of genome-wide association studies,” Nature Reviews Genetics, vol. 20, p. 467–484, 2019. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources