

Leveraging epigenomes and three-dimensional genome organization for interpreting regulatory variation
- PMID: 37428809
- PMCID: PMC10358954
- DOI: 10.1371/journal.pcbi.1011286
Leveraging epigenomes and three-dimensional genome organization for interpreting regulatory variation
Abstract
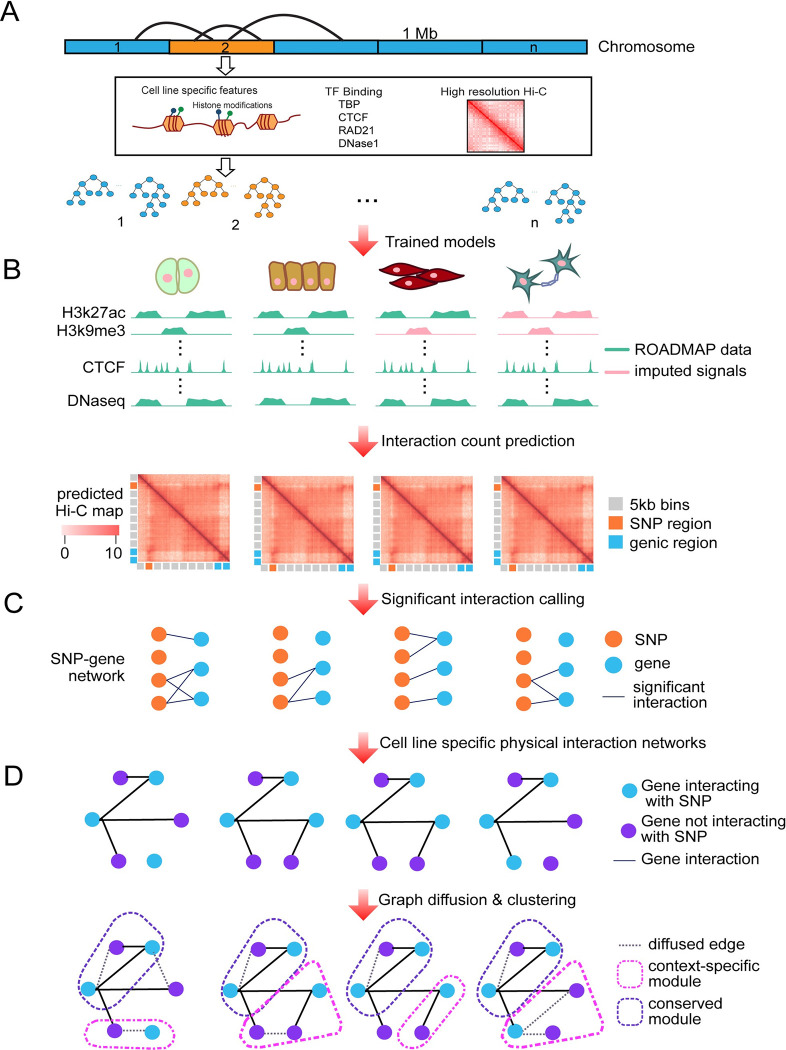
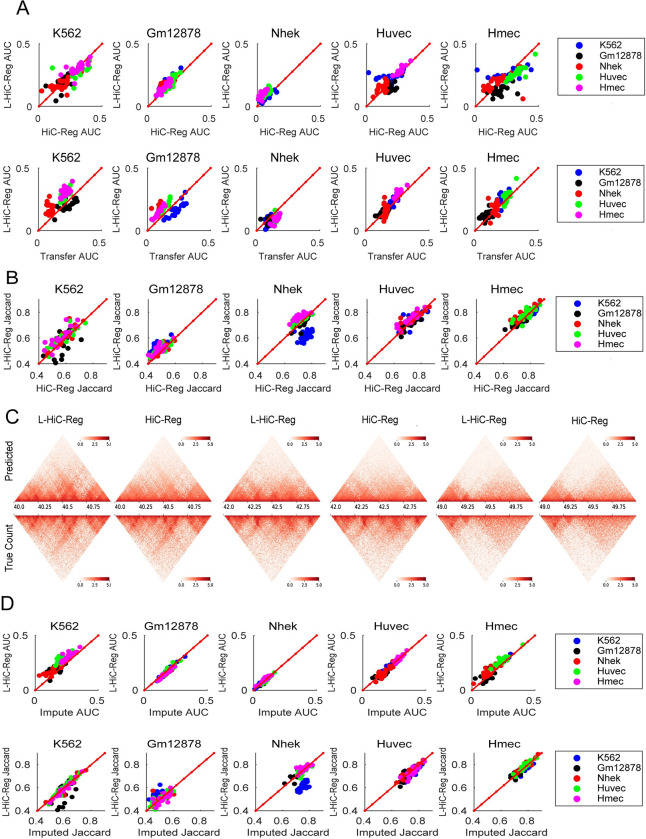
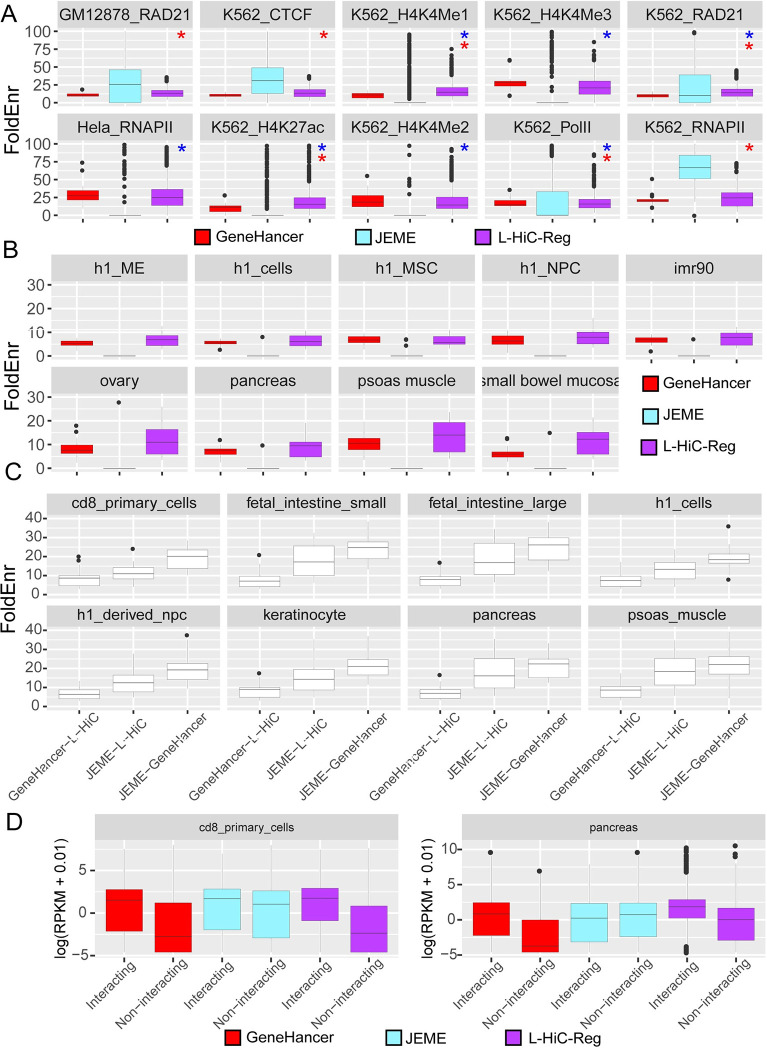
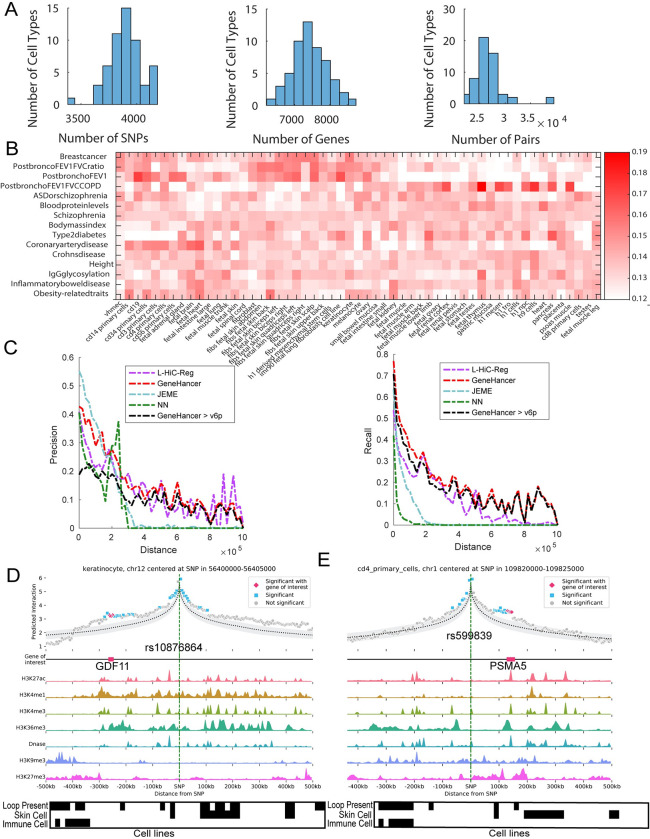
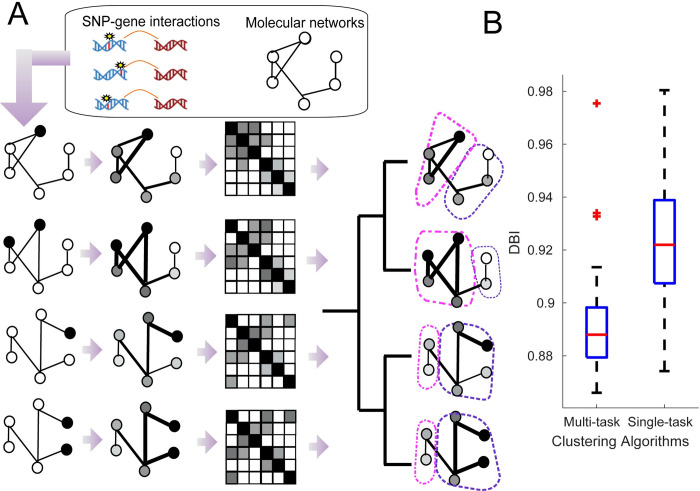
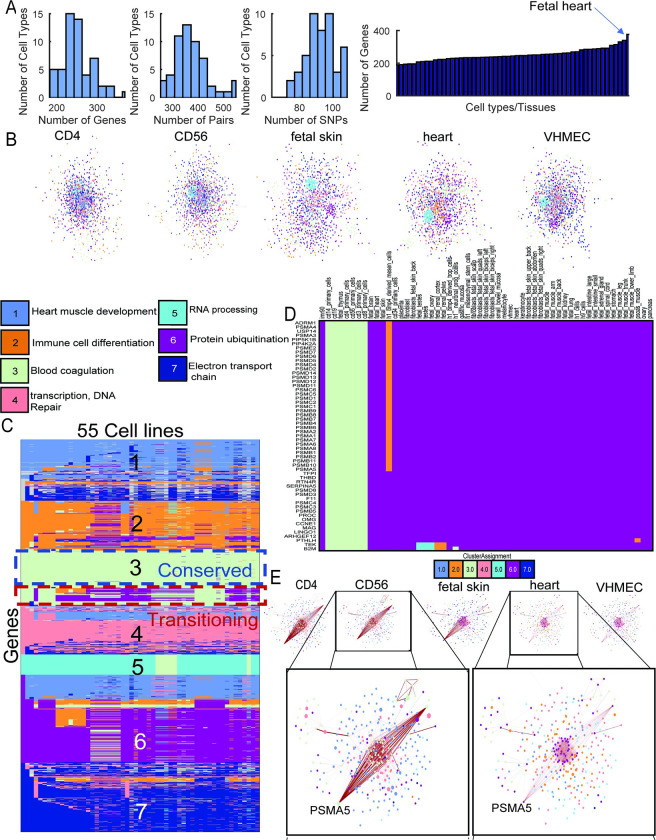
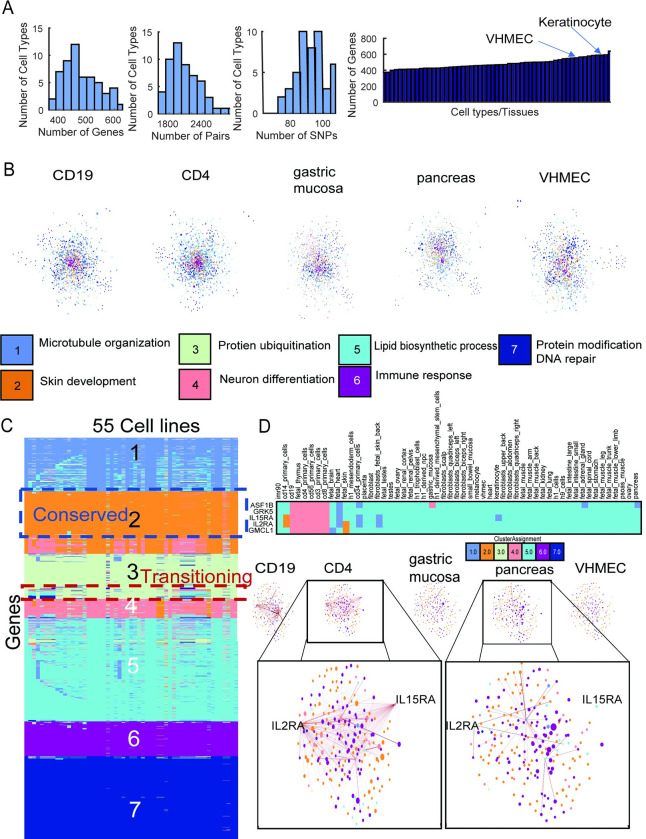
Understanding the impact of regulatory variants on complex phenotypes is a significant challenge because the genes and pathways that are targeted by such variants and the cell type context in which regulatory variants operate are typically unknown. Cell-type-specific long-range regulatory interactions that occur between a distal regulatory sequence and a gene offer a powerful framework for examining the impact of regulatory variants on complex phenotypes. However, high-resolution maps of such long-range interactions are available only for a handful of cell types. Furthermore, identifying specific gene subnetworks or pathways that are targeted by a set of variants is a significant challenge. We have developed L-HiC-Reg, a Random Forests regression method to predict high-resolution contact counts in new cell types, and a network-based framework to identify candidate cell-type-specific gene networks targeted by a set of variants from a genome-wide association study (GWAS). We applied our approach to predict interactions in 55 Roadmap Epigenomics Mapping Consortium cell types, which we used to interpret regulatory single nucleotide polymorphisms (SNPs) in the NHGRI-EBI GWAS catalogue. Using our approach, we performed an in-depth characterization of fifteen different phenotypes including schizophrenia, coronary artery disease (CAD) and Crohn's disease. We found differentially wired subnetworks consisting of known as well as novel gene targets of regulatory SNPs. Taken together, our compendium of interactions and the associated network-based analysis pipeline leverages long-range regulatory interactions to examine the context-specific impact of regulatory variation in complex phenotypes.
Copyright: © 2023 Baur et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
