

Complement and platelets: prothrombotic cell activation requires membrane attack complex-induced release of danger signals
- PMID: 37428869
- PMCID: PMC10625899
- DOI: 10.1182/bloodadvances.2023010817
Complement and platelets: prothrombotic cell activation requires membrane attack complex-induced release of danger signals
Abstract
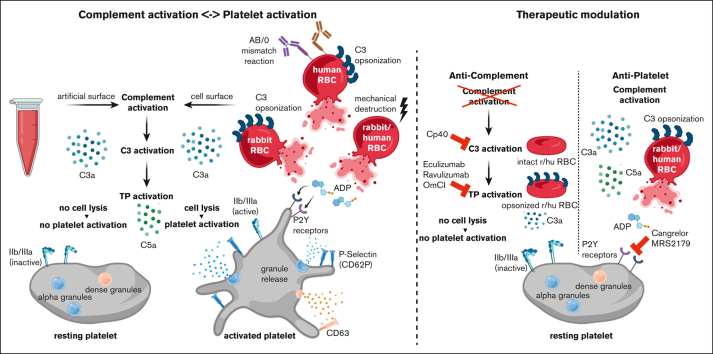
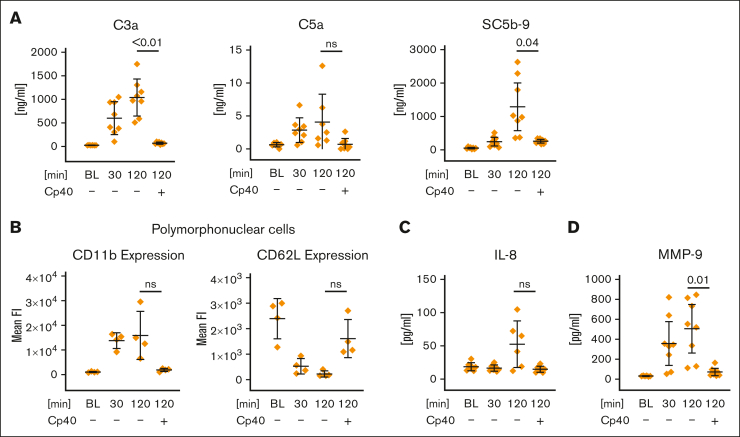
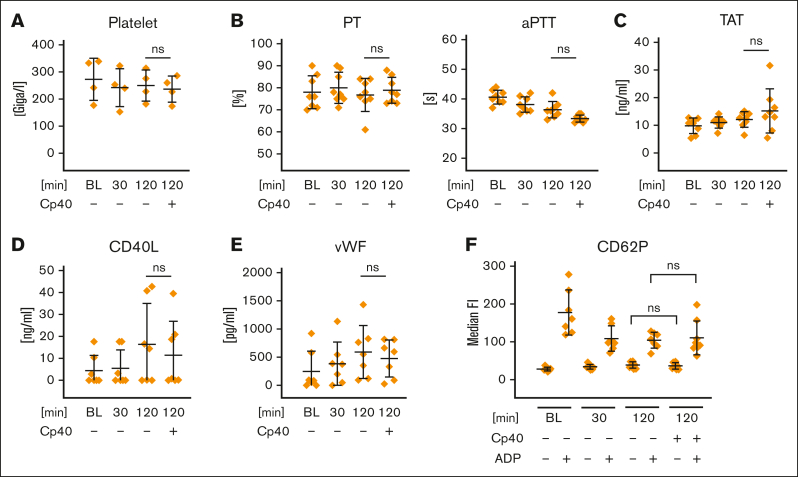
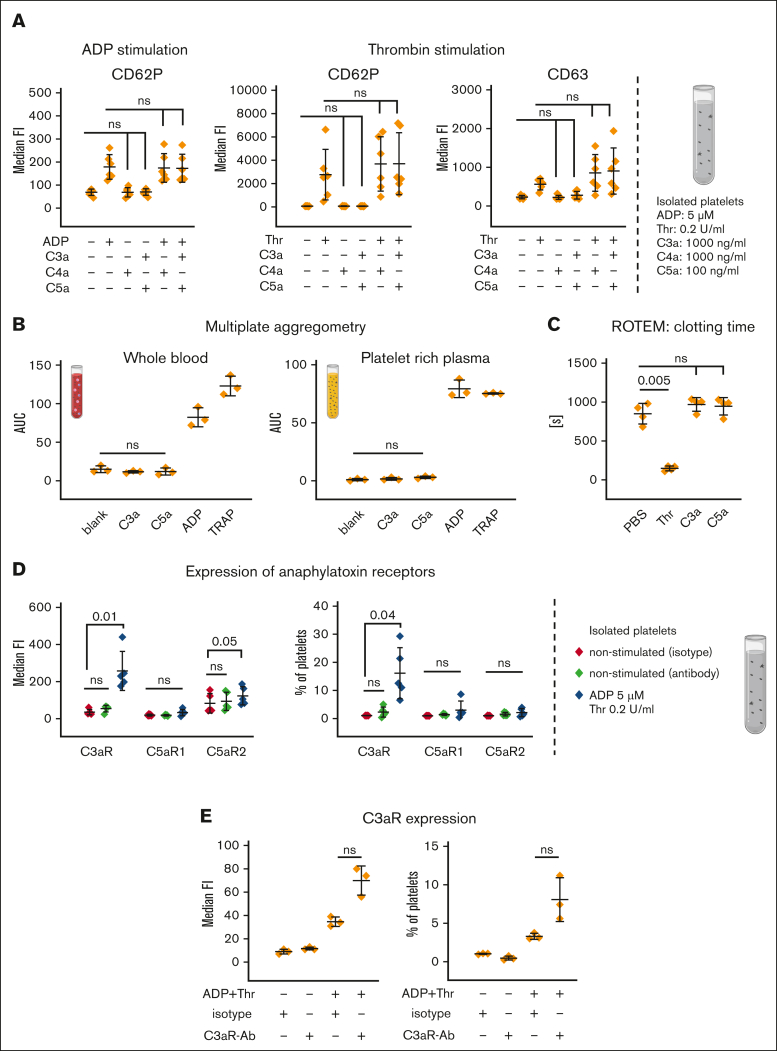
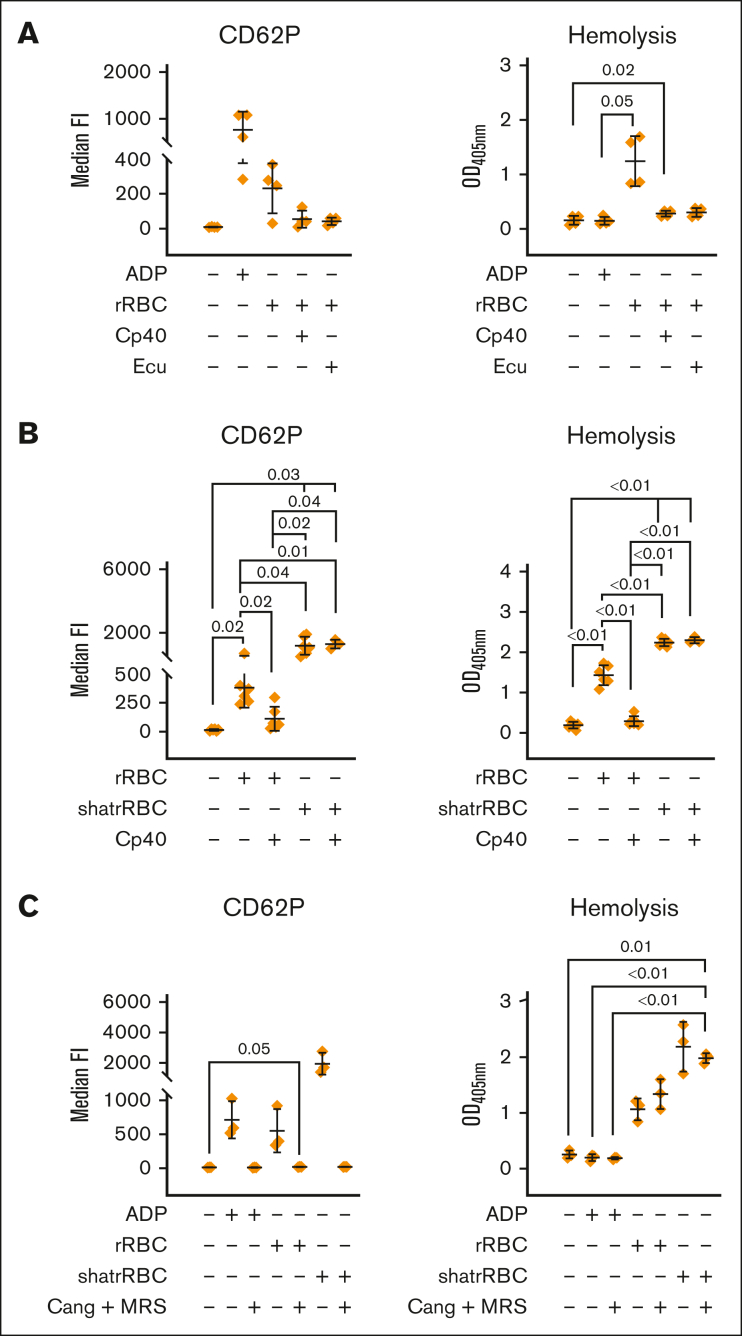
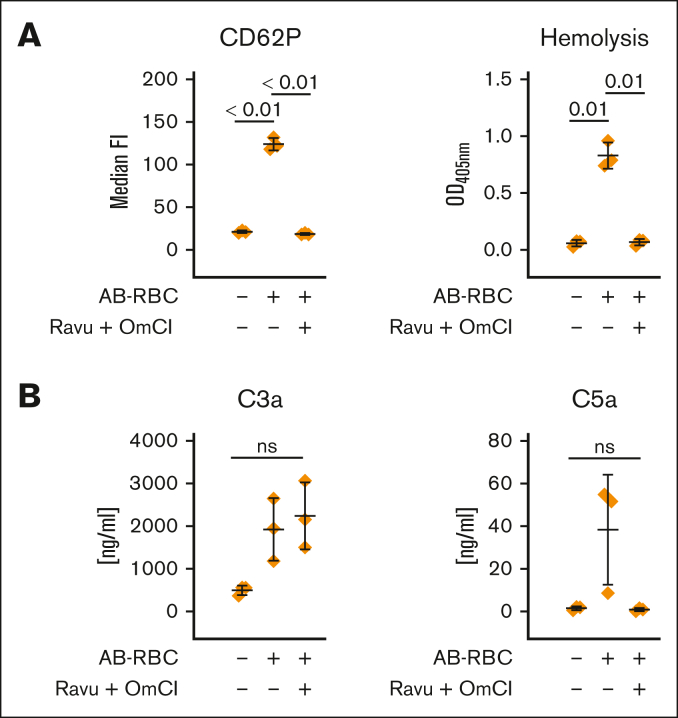
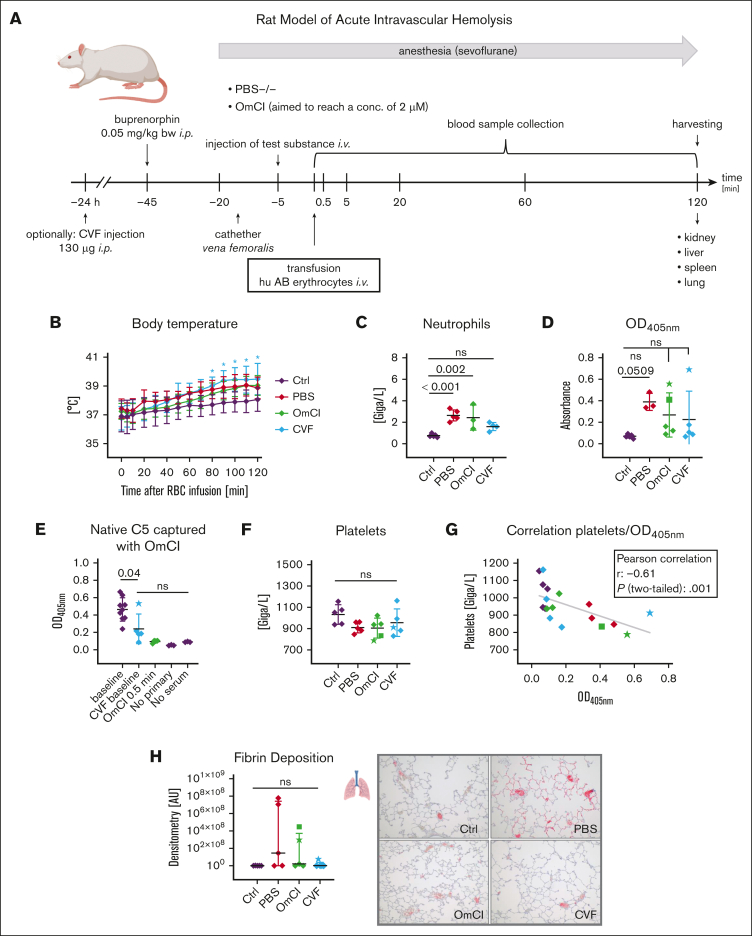
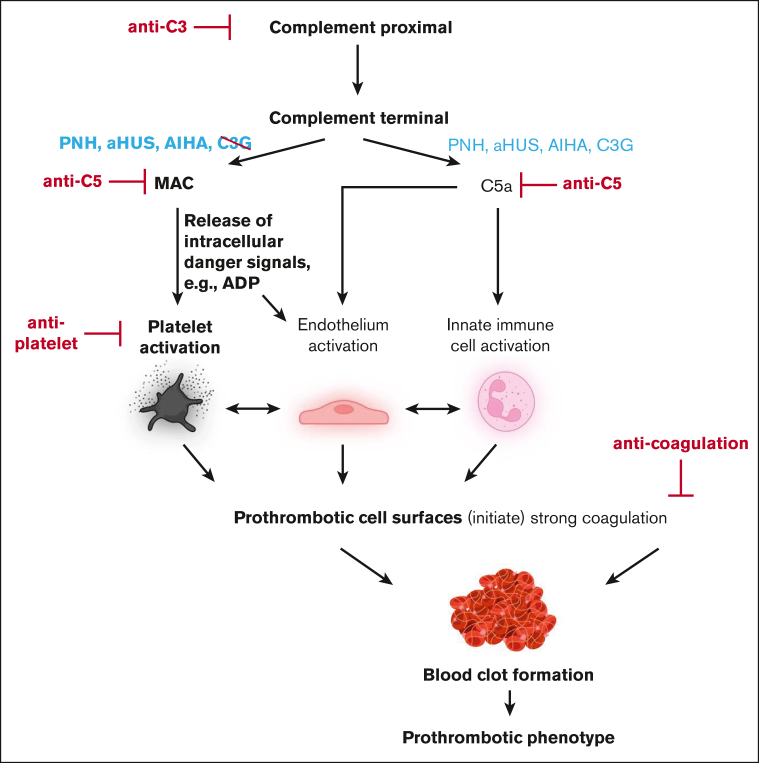
Complement activation in the diseases paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS) results in cytolysis and fatal thrombotic events, which are largely refractory to anticoagulation and/or antiplatelet therapy. Anticomplement therapy, however, efficiently prevents thrombotic events in PNH and aHUS, but the underlying mechanisms remain unresolved. We show that complement-mediated hemolysis in whole blood induces platelet activation similarly to activation by adenosine 5'-diphosphate (ADP). Blockage of C3 or C5 abolished platelet activation. We found that human platelets failed to respond functionally to the anaphylatoxins C3a and C5a. Instead, complement activation did lead to prothrombotic cell activation in the whole blood when membrane attack complex (MAC)-mediated cytolysis occurred. Consequently, we demonstrate that ADP receptor antagonists efficiently inhibited platelet activation, although full complement activation, which causes hemolysis, occurred. By using an established model of mismatched erythrocyte transfusions in rats, we crossvalidated these findings in vivo using the complement inhibitor OmCI and cobra venom factor. Consumptive complement activation in this animal model only led to a thrombotic phenotype when MAC-mediated cytolysis occurred. In conclusion, complement activation only induces substantial prothrombotic cell activation if terminal pathway activation culminates in MAC-mediated release of intracellular ADP. These results explain why anticomplement therapy efficiently prevents thromboembolisms without interfering negatively with hemostasis.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: C.Q.S., H.S., and M.H.-L. are listed as inventors on (a) patent application(s) that describes the use of complement inhibitors for therapeutic applications. C.Q.S. has received research funding from pharmaceutical companies. M.H.-L., C.Q.S., and H.S. received honoraria for speaking at symposia organized by Alexion Pharmaceuticals. H.S. served on advisory committees for Alexion AstraZeneca Rare Diseases, Sanofi, Sobi, and Novartis; received research funding from Alexion Pharmaceuticals (all to the University of Ulm); and served on an advisory committee for Ra Pharmaceuticals and Alnylam. The remaining authors declare no competing financial interests.
Figures
Comment in
-
ADP: the missing link between thrombosis and hemolysis.Blood Adv. 2023 Oct 24;7(20):6364-6366. doi: 10.1182/bloodadvances.2023011186. Blood Adv. 2023. PMID: 37874560 Free PMC article. No abstract available.
References
-
- Luzzatto L, Gianfaldoni G, Notaro R. Management of paroxysmal nocturnal haemoglobinuria: a personal view. Br J Haematol. 2011;153(6):709–720. - PubMed
-
- Parker CJ. The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2007;35(4):523–533. - PubMed
-
- Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985–4996. quiz 5105. - PubMed
-
- Audebert HJ, Planck J, Eisenburg M, Schrezenmeier H, Haberl RL. Cerebral ischemic infarction in paroxysmal nocturnal hemoglobinuria report of 2 cases and updated review of 7 previously published patients. J Neurol. 2005;252(11):1379–1386. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
