

Individual bat virome analysis reveals co-infection and spillover among bats and virus zoonotic potential
- PMID: 37429936
- PMCID: PMC10333379
- DOI: 10.1038/s41467-023-39835-1
Individual bat virome analysis reveals co-infection and spillover among bats and virus zoonotic potential
Abstract
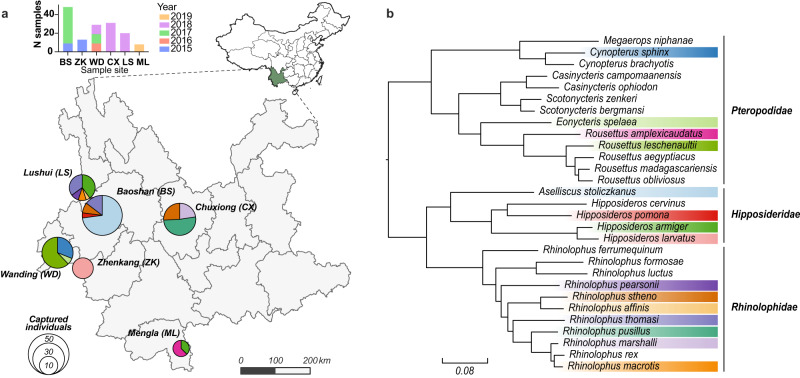
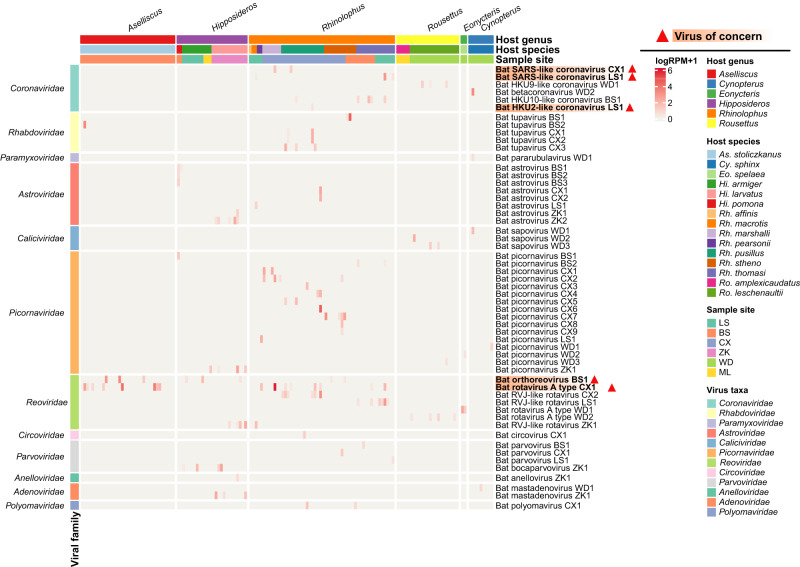
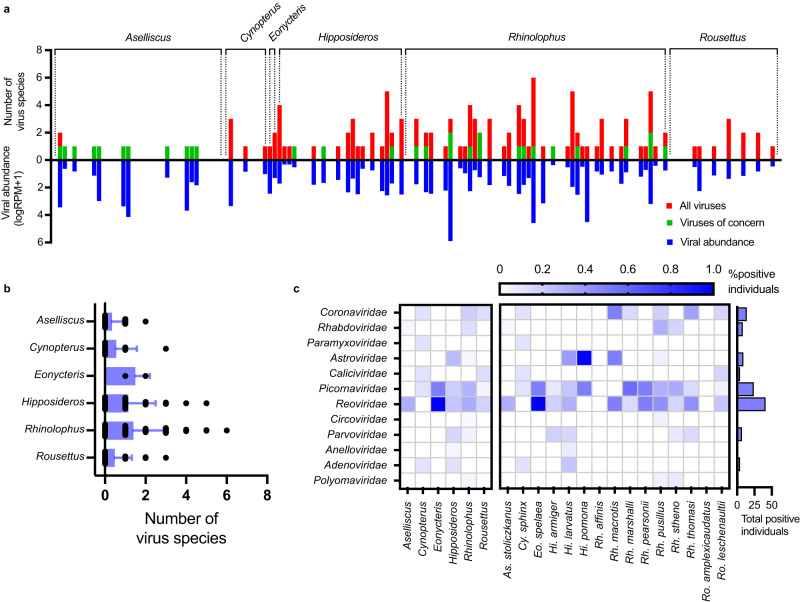
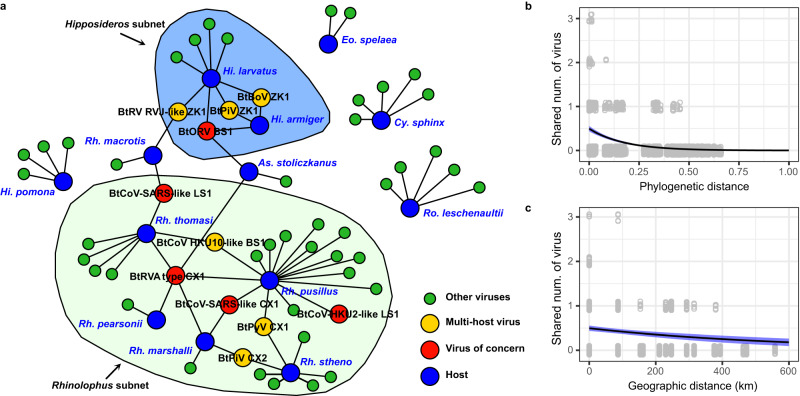
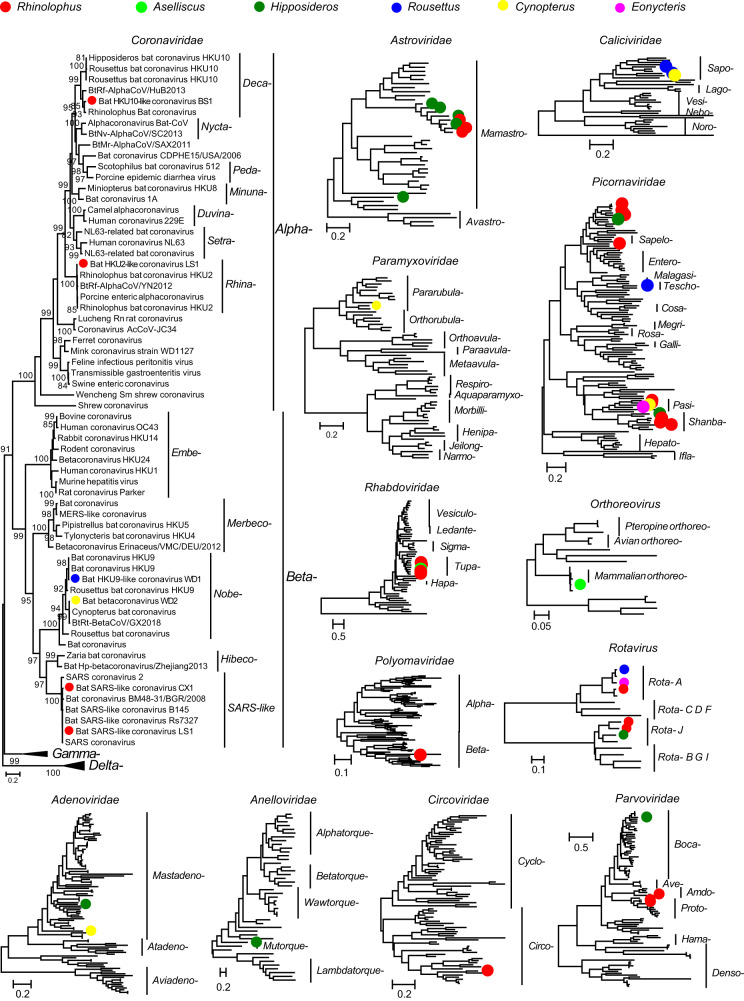
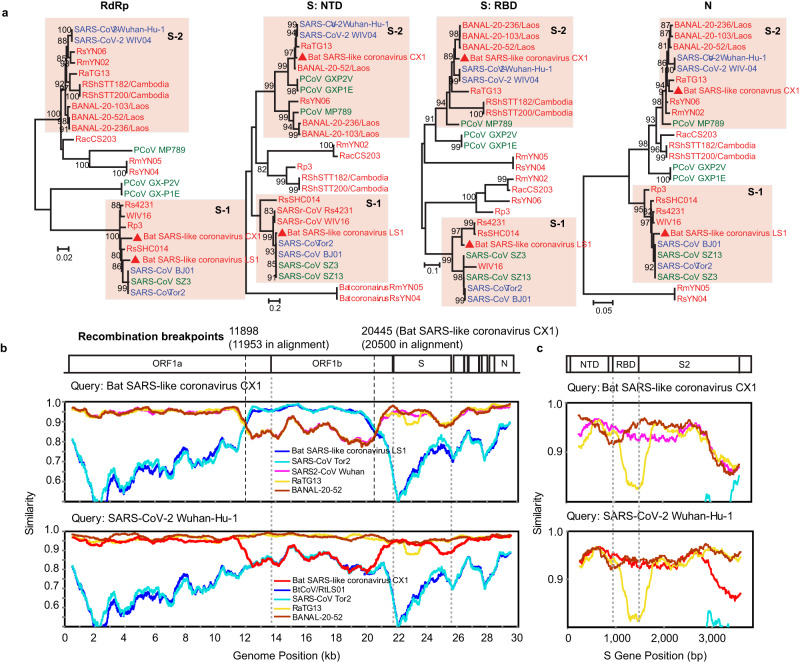
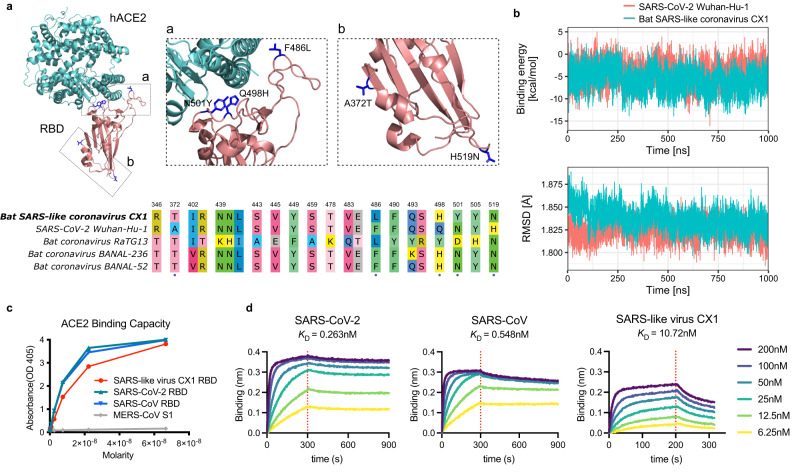
Bats are reservoir hosts for many zoonotic viruses. Despite this, relatively little is known about the diversity and abundance of viruses within individual bats, and hence the frequency of virus co-infection and spillover among them. We characterize the mammal-associated viruses in 149 individual bats sampled from Yunnan province, China, using an unbiased meta-transcriptomics approach. This reveals a high frequency of virus co-infection (simultaneous infection of bat individuals by multiple viral species) and spillover among the animals studied, which may in turn facilitate virus recombination and reassortment. Of note, we identify five viral species that are likely to be pathogenic to humans or livestock, based on phylogenetic relatedness to known pathogens or in vitro receptor binding assays. This includes a novel recombinant SARS-like coronavirus that is closely related to both SARS-CoV and SARS-CoV-2. In vitro assays indicate that this recombinant virus can utilize the human ACE2 receptor such that it is likely to be of increased emergence risk. Our study highlights the common occurrence of co-infection and spillover of bat viruses and their implications for virus emergence.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
