

Ethnicity, consanguinity, and genetic architecture of hypertrophic cardiomyopathy
- PMID: 37431535
- PMCID: PMC10733735
- DOI: 10.1093/eurheartj/ehad372
Ethnicity, consanguinity, and genetic architecture of hypertrophic cardiomyopathy
Abstract
Aims: Hypertrophic cardiomyopathy (HCM) is characterized by phenotypic heterogeneity that is partly explained by the diversity of genetic variants contributing to disease. Accurate interpretation of these variants constitutes a major challenge for diagnosis and implementing precision medicine, especially in understudied populations. The aim is to define the genetic architecture of HCM in North African cohorts with high consanguinity using ancestry-matched cases and controls.
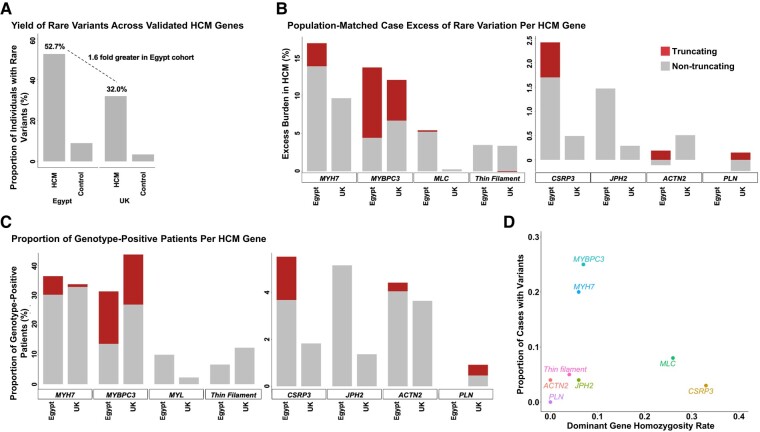
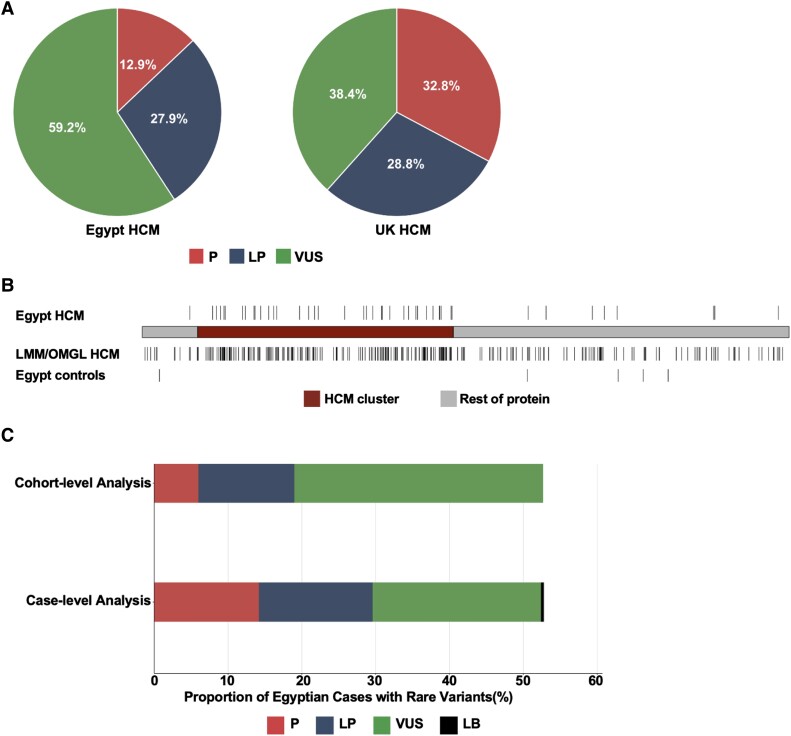
Methods and results: Prospective Egyptian patients (n = 514) and controls (n = 400) underwent clinical phenotyping and genetic testing. Rare variants in 13 validated HCM genes were classified according to standard clinical guidelines and compared with a prospective HCM cohort of majority European ancestry (n = 684). A higher prevalence of homozygous variants was observed in Egyptian patients (4.1% vs. 0.1%, P = 2 × 10-7), with variants in the minor HCM genes MYL2, MYL3, and CSRP3 more likely to present in homozygosity than the major genes, suggesting these variants are less penetrant in heterozygosity. Biallelic variants in the recessive HCM gene TRIM63 were detected in 2.1% of patients (five-fold greater than European patients), highlighting the importance of recessive inheritance in consanguineous populations. Finally, rare variants in Egyptian HCM patients were less likely to be classified as (likely) pathogenic compared with Europeans (40.8% vs. 61.6%, P = 1.6 × 10-5) due to the underrepresentation of Middle Eastern populations in current reference resources. This proportion increased to 53.3% after incorporating methods that leverage new ancestry-matched controls presented here.
Conclusion: Studying consanguineous populations reveals novel insights with relevance to genetic testing and our understanding of the genetic architecture of HCM.
Keywords: Egyptian collaborative cardiac genomics project; Homozygosity; NGS.
© The Author(s) 2023. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures

Comment in
-
The moral and practical urgency of increasing diversity in genomics.Eur Heart J. 2023 Dec 21;44(48):5157-5159. doi: 10.1093/eurheartj/ehad365. Eur Heart J. 2023. PMID: 37377076 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
