

Metagenomic Surveillance of Viral Gastroenteritis in a Public Health Setting
- PMID: 37432120
- PMCID: PMC10434279
- DOI: 10.1128/spectrum.05022-22
Metagenomic Surveillance of Viral Gastroenteritis in a Public Health Setting
Abstract
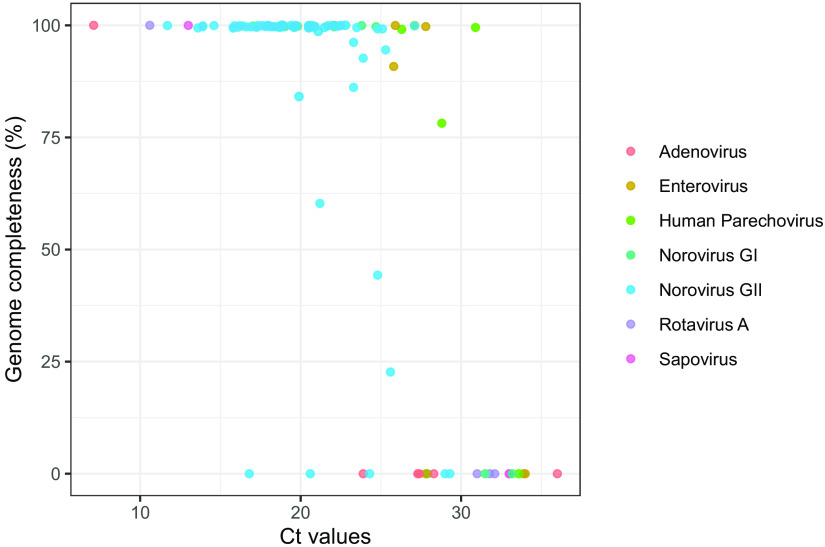
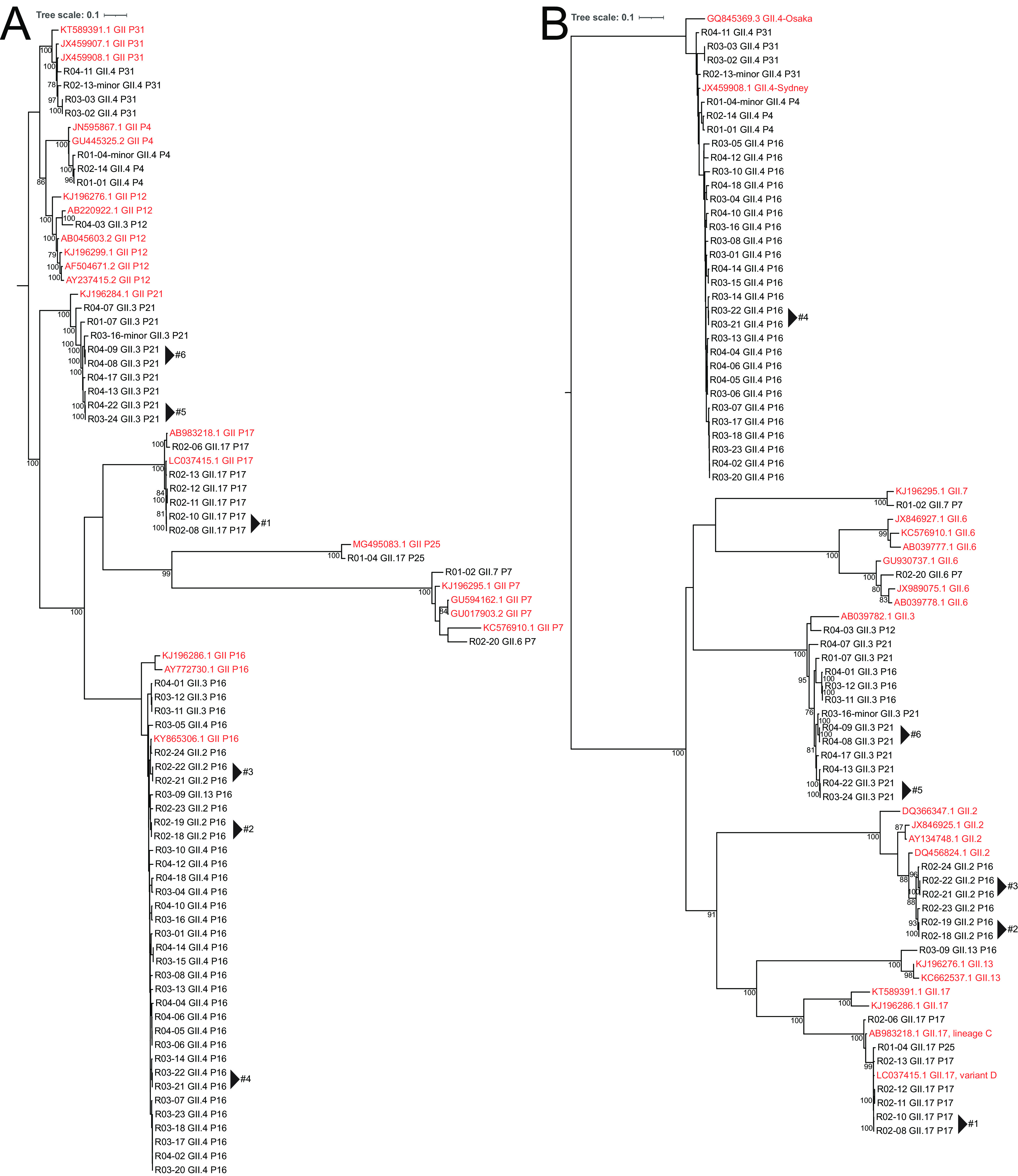
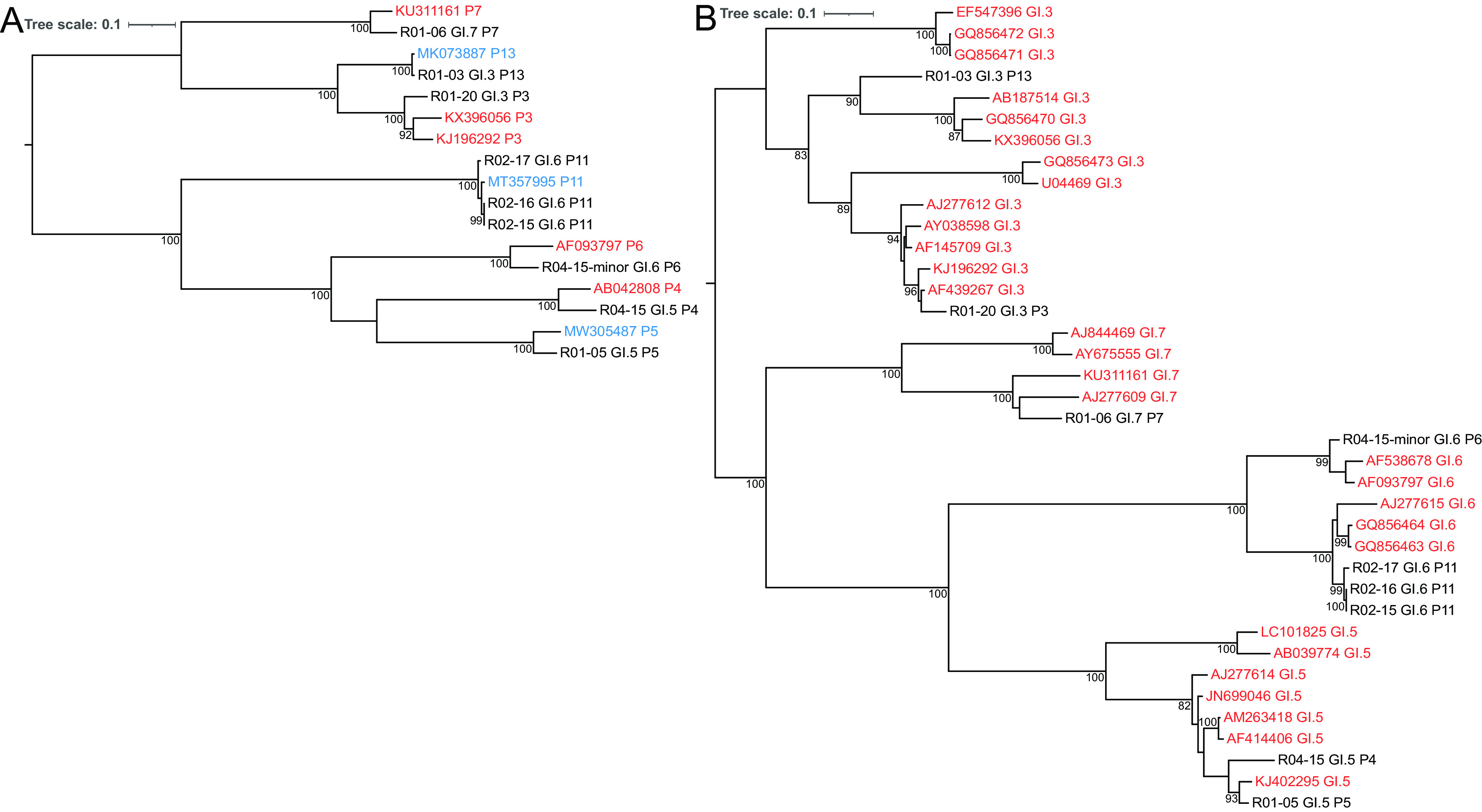
Norovirus is the primary cause of viral gastroenteritis (GE). To investigate norovirus epidemiology, there is a need for whole-genome sequencing and reference sets consisting of complete genomes. To investigate the potential of shotgun metagenomic sequencing on the Illumina platform for whole-genome sequencing, 71 reverse transcriptase quantitative PCR (RT-qPCR) norovirus positive-feces (threshold cycle [CT], <30) samples from norovirus surveillance within The Netherlands were subjected to metagenomic sequencing. Data were analyzed through an in-house next-generation sequencing (NGS) analysis workflow. Additionally, we assessed the potential of metagenomic sequencing for the surveillance of off-target viruses that are of importance for public health, e.g., sapovirus, rotavirus A, enterovirus, parechovirus, aichivirus, adenovirus, and bocaparvovirus. A total of 60 complete and 10 partial norovirus genomes were generated, representing 7 genogroup I capsid genotypes and 12 genogroup II capsid genotypes. In addition to the norovirus genomes, the metagenomic approach yielded partial or complete genomes of other viruses for 39% of samples from children and 6.7% of samples from adults, including adenovirus 41 (N = 1); aichivirus 1 (N = 1); coxsackievirus A2 (N = 2), A4 (N = 2), A5 (N = 1), and A16 (N = 1); bocaparvovirus 1 (N = 1) and 3 (N = 1); human parechovirus 1 (N = 2) and 3 (N = 1); Rotavirus A (N = 1); and a sapovirus GI.7 (N = 1). The sapovirus GI.7 was initially not detected through RT-qPCR and warranted an update of the primer and probe set. Metagenomic sequencing on the Illumina platform robustly determines complete norovirus genomes and may be used to broaden gastroenteritis surveillance by capturing off-target enteric viruses. IMPORTANCE Viral gastroenteritis results in significant morbidity and mortality in vulnerable individuals and is primarily caused by norovirus. To investigate norovirus epidemiology, there is a need for whole-genome sequencing and reference sets consisting of full genomes. Using surveillance samples sent to the Dutch National Institute for Public Health and the Environment (RIVM), we compared metagenomics against conventional techniques, such as RT-qPCR and Sanger-sequencing, with norovirus as the target pathogen. We determined that metagenomics is a robust method to generate complete norovirus genomes, in parallel to many off-target pathogenic enteric virus genomes, thereby broadening our surveillance efforts. Moreover, we detected a sapovirus that was not detected by our validated gastroenteritis RT-qPCR panel, which exemplifies the strength of metagenomics. Our study shows that metagenomics can be used for public health gastroenteritis surveillance, the generation of reference-sets for molecular epidemiology, and how it compares to current surveillance strategies.
Keywords: enteric viruses; gastroenteritis; next-generation sequencing; norovirus; public health; surveillance; surveillance studies; transmissible gastroenteritis virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Havelaar AH, Kirk MD, Torgerson PR, Gibb HJ, Hald T, Lake RJ, Praet N, Bellinger DC, de Silva NR, Gargouri N, Speybroeck N, Cawthorne A, Mathers C, Stein C, Angulo FJ, Devleesschauwer B, Adegoke GO, Afshari R, Alasfoor D, Baines J, Balakrishnan K, Hamza Wb, Black RE, Bolger PM, Chaicumpa W, Cravioto A, Döpfer D, Ehiri JE, Fazil A, Ferreccio C, Fèvre EM, Hall G, Kasuga F, Keddy KH, Lanata CF, Lei H, Liu X, Manyindo B, Nasinyama G, Ongolo-Zogo P, Pitt JI, Rokni MB, Sripa B, van Leeuwen R, Verger P, Willingham AL, Zhou XN, Aspinall W, Buchanan R, Budke C, on behalf of World Health Organization Foodborne Disease Burden Epidemiology Reference Group , et al. 2015. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med 12:e1001923. doi:10.1371/journal.pmed.1001923. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
