

Uncovering Footprints of Natural Selection Through Spectral Analysis of Genomic Summary Statistics
- PMID: 37433019
- PMCID: PMC10365025
- DOI: 10.1093/molbev/msad157
Uncovering Footprints of Natural Selection Through Spectral Analysis of Genomic Summary Statistics
Abstract
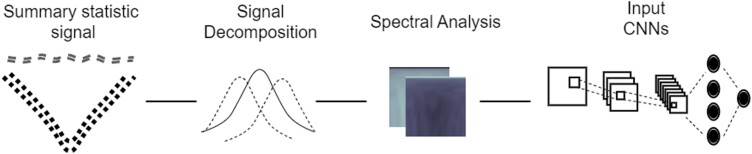
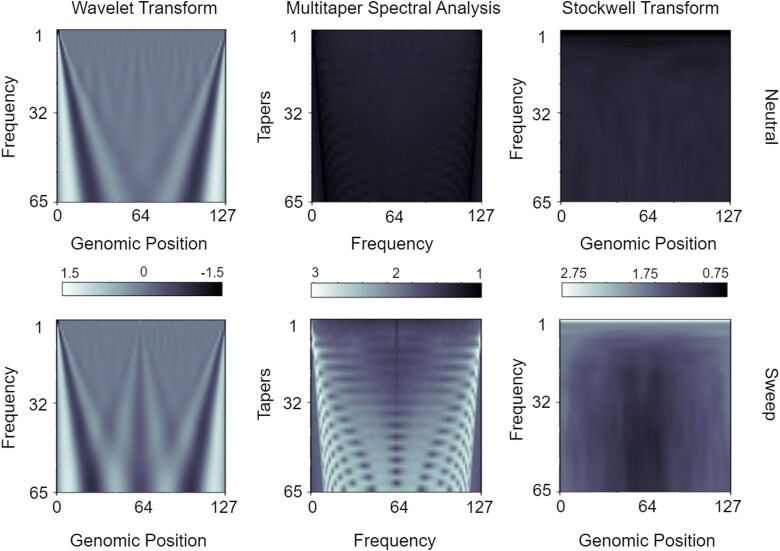
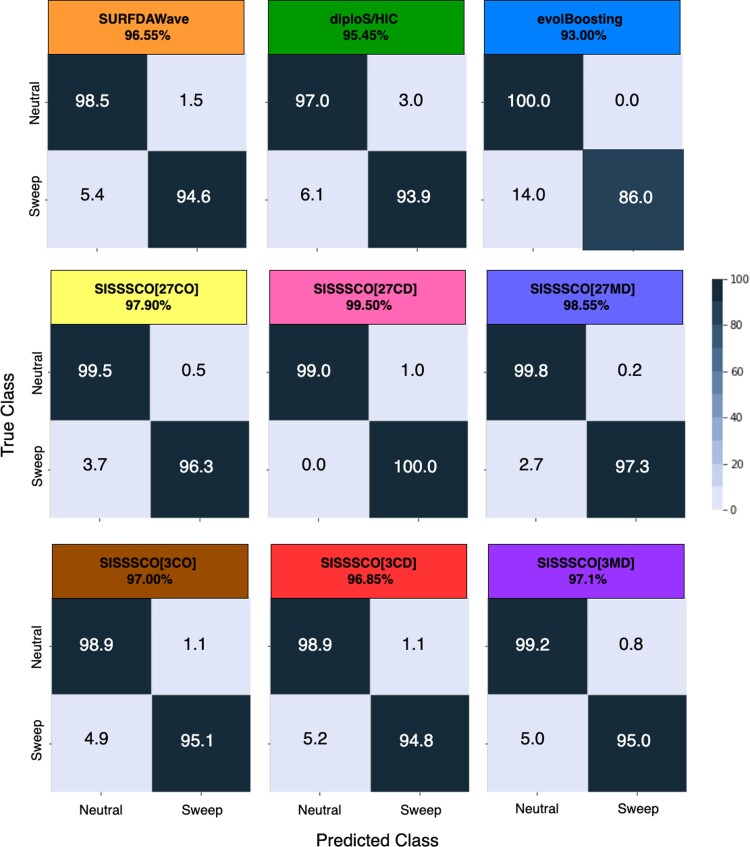
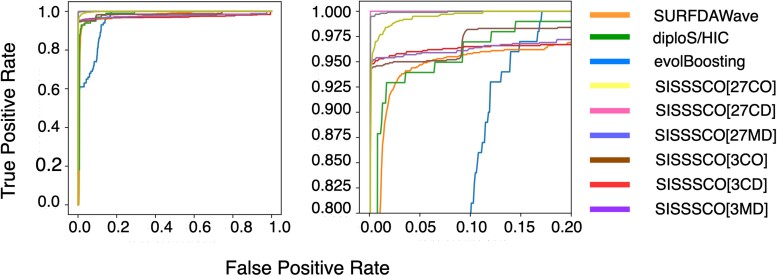
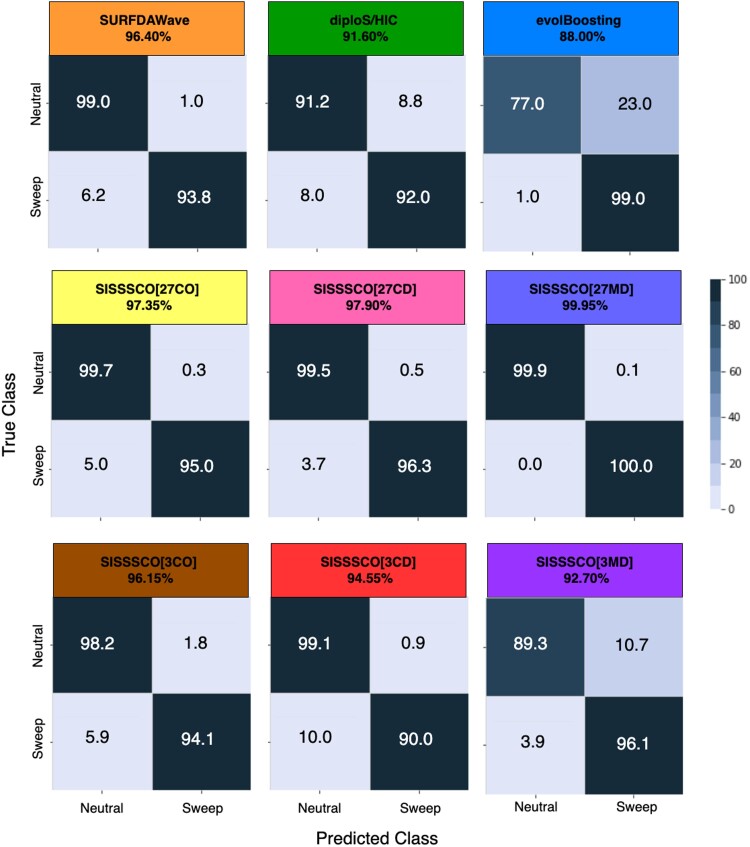
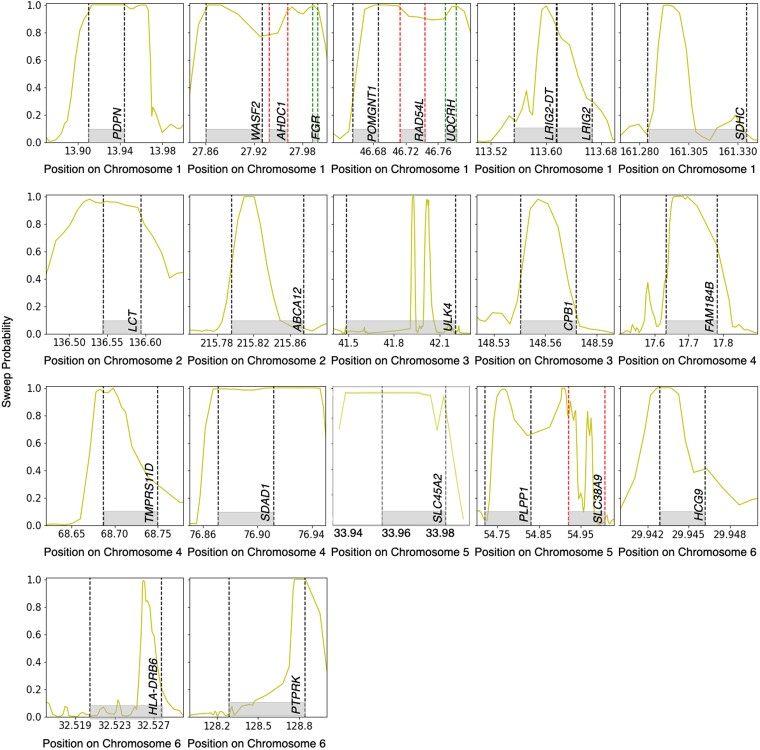
Natural selection leaves a spatial pattern along the genome, with a haplotype distribution distortion near the selected locus that fades with distance. Evaluating the spatial signal of a population-genetic summary statistic across the genome allows for patterns of natural selection to be distinguished from neutrality. Considering the genomic spatial distribution of multiple summary statistics is expected to aid in uncovering subtle signatures of selection. In recent years, numerous methods have been devised that consider genomic spatial distributions across summary statistics, utilizing both classical machine learning and deep learning architectures. However, better predictions may be attainable by improving the way in which features are extracted from these summary statistics. We apply wavelet transform, multitaper spectral analysis, and S-transform to summary statistic arrays to achieve this goal. Each analysis method converts one-dimensional summary statistic arrays to two-dimensional images of spectral analysis, allowing simultaneous temporal and spectral assessment. We feed these images into convolutional neural networks and consider combining models using ensemble stacking. Our modeling framework achieves high accuracy and power across a diverse set of evolutionary settings, including population size changes and test sets of varying sweep strength, softness, and timing. A scan of central European whole-genome sequences recapitulated well-established sweep candidates and predicted novel cancer-associated genes as sweeps with high support. Given that this modeling framework is also robust to missing genomic segments, we believe that it will represent a welcome addition to the population-genomic toolkit for learning about adaptive processes from genomic data.
Keywords: artificial intelligence; natural selection; signal decomposition.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures


References
-
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al. 2015. TensorFlow: large-scale machine learning on heterogeneous systems. Available from:https://www.tensorflow.org/
-
- Abu-Mostafa YS, Atiya AF. 1996. Introduction to financial forecasting. Appl Intel. 6:205–213.
-
- Agrawal A, Mittal N. 2020. Using CNN for facial expression recognition: a study of the effects of kernel size and number of filters on accuracy. Vis Comput. 36:405–412.
-
- Akiyama M. 2014. The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim Biophys Acta. 1841:435–440. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
