

Role of neuroinflammation in neurodegeneration development
- PMID: 37433768
- PMCID: PMC10336149
- DOI: 10.1038/s41392-023-01486-5
Role of neuroinflammation in neurodegeneration development
Abstract
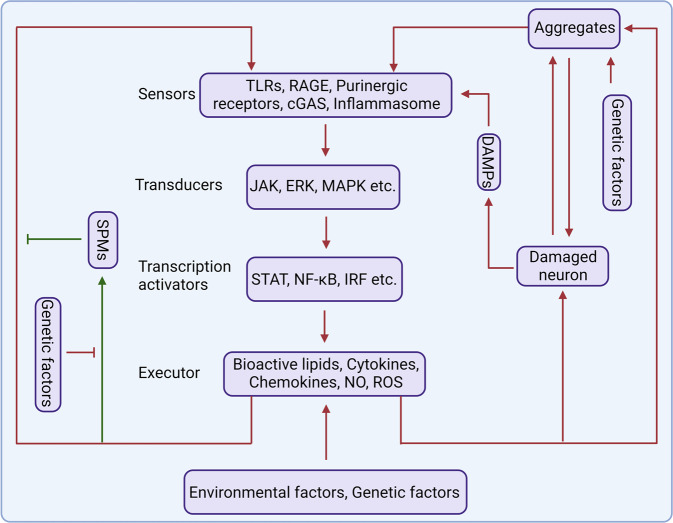
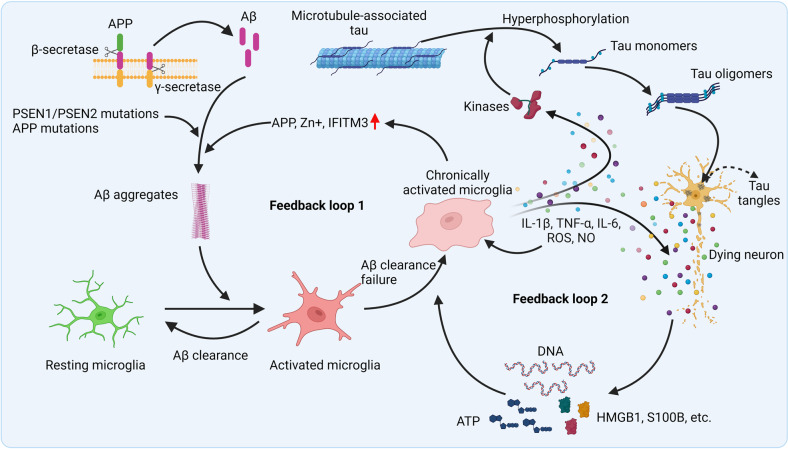
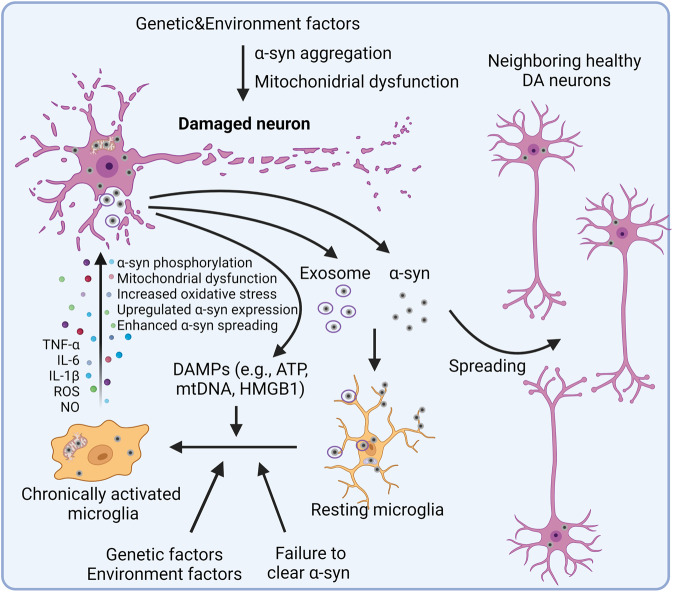
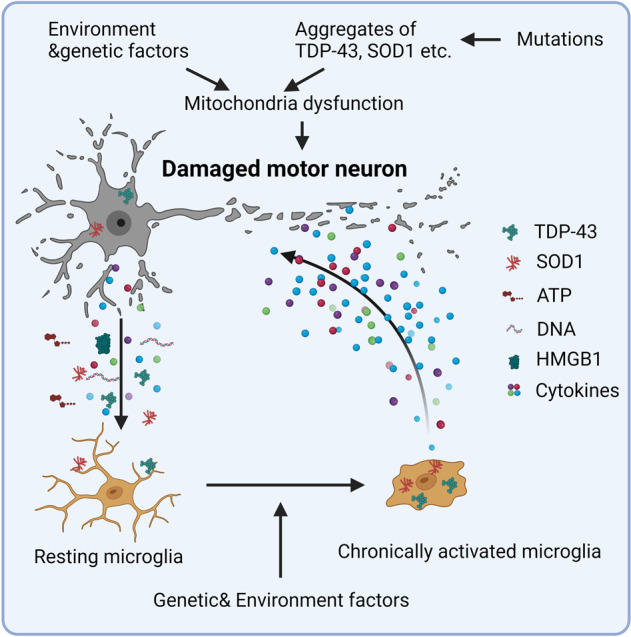
Studies in neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease and Amyotrophic lateral sclerosis, Huntington's disease, and so on, have suggested that inflammation is not only a result of neurodegeneration but also a crucial player in this process. Protein aggregates which are very common pathological phenomenon in neurodegeneration can induce neuroinflammation which further aggravates protein aggregation and neurodegeneration. Actually, inflammation even happens earlier than protein aggregation. Neuroinflammation induced by genetic variations in CNS cells or by peripheral immune cells may induce protein deposition in some susceptible population. Numerous signaling pathways and a range of CNS cells have been suggested to be involved in the pathogenesis of neurodegeneration, although they are still far from being completely understood. Due to the limited success of traditional treatment methods, blocking or enhancing inflammatory signaling pathways involved in neurodegeneration are considered to be promising strategies for the therapy of neurodegenerative diseases, and many of them have got exciting results in animal models or clinical trials. Some of them, although very few, have been approved by FDA for clinical usage. Here we comprehensively review the factors affecting neuroinflammation and the major inflammatory signaling pathways involved in the pathogenicity of neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and Amyotrophic lateral sclerosis. We also summarize the current strategies, both in animal models and in the clinic, for the treatment of neurodegenerative diseases.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
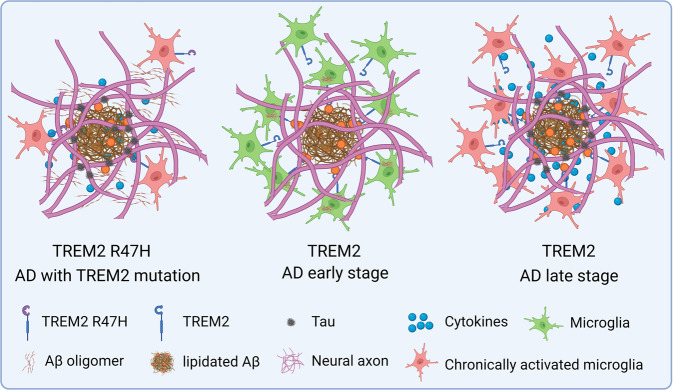
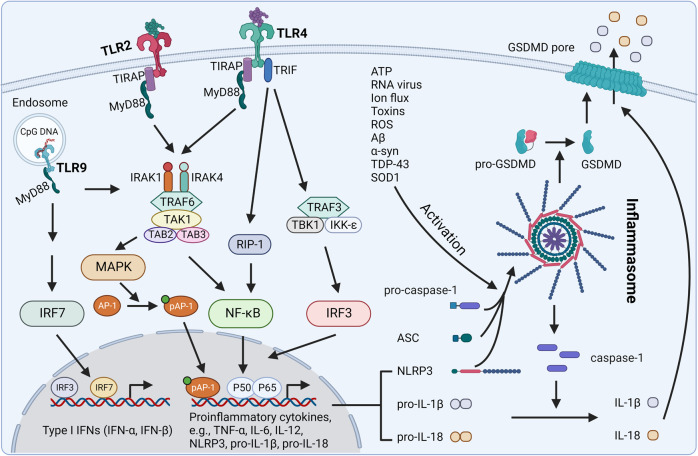
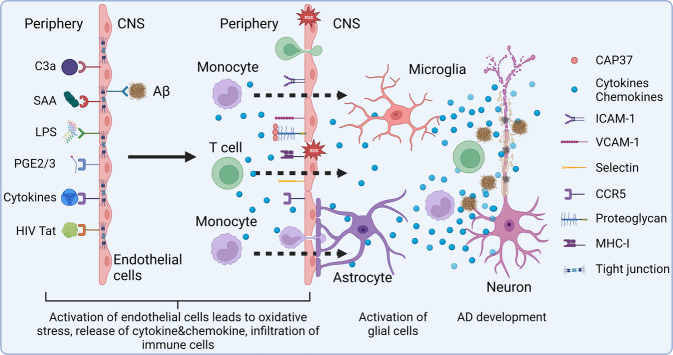
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
