

Spatial cellular architecture predicts prognosis in glioblastoma
- PMID: 37433817
- PMCID: PMC10336135
- DOI: 10.1038/s41467-023-39933-0
Spatial cellular architecture predicts prognosis in glioblastoma
Abstract
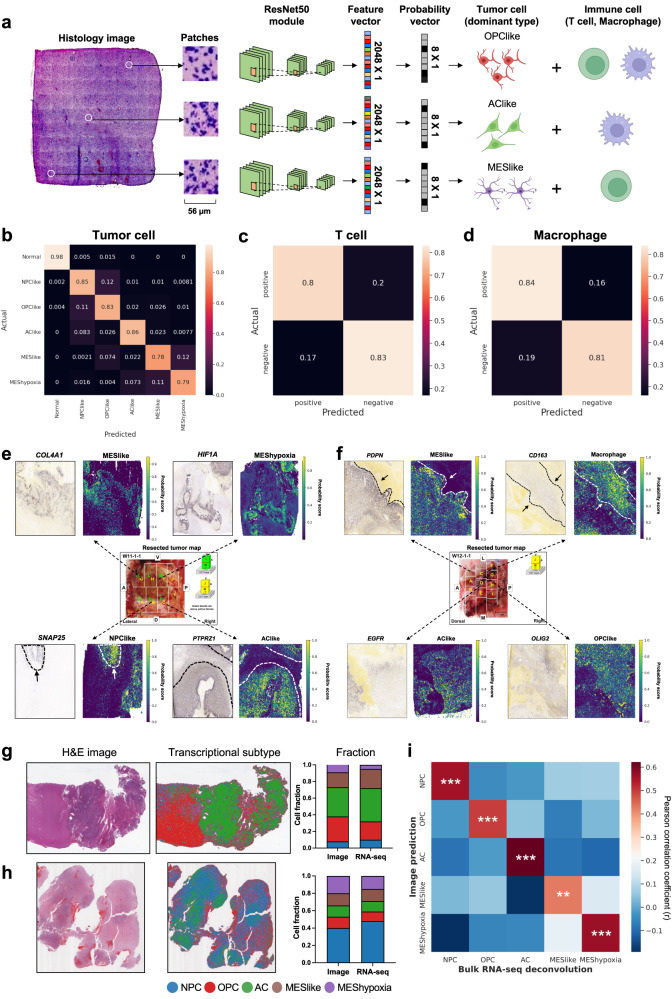
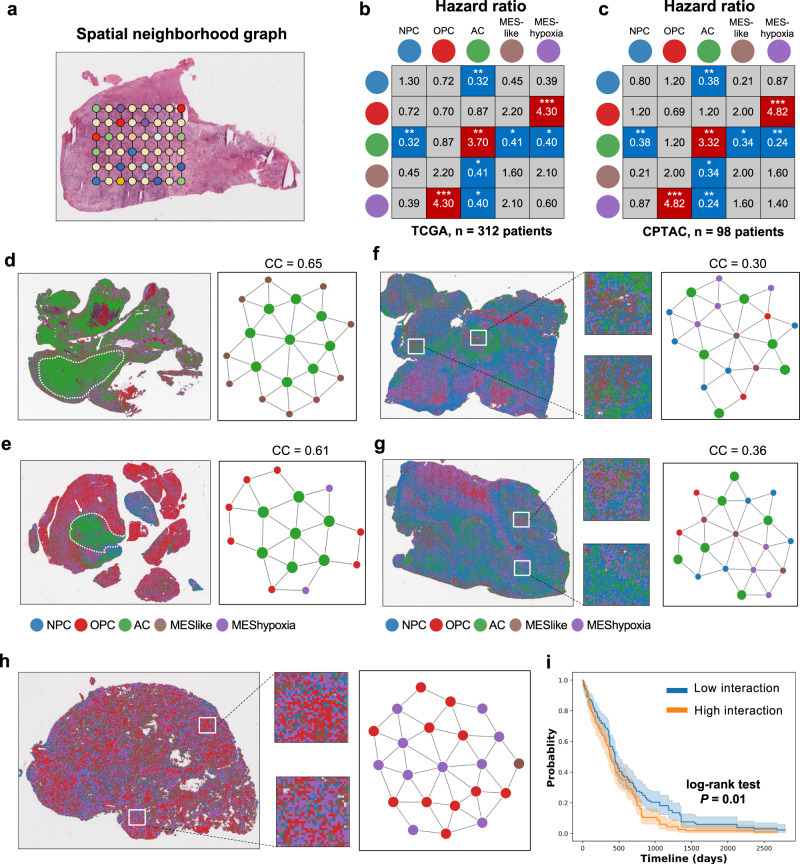
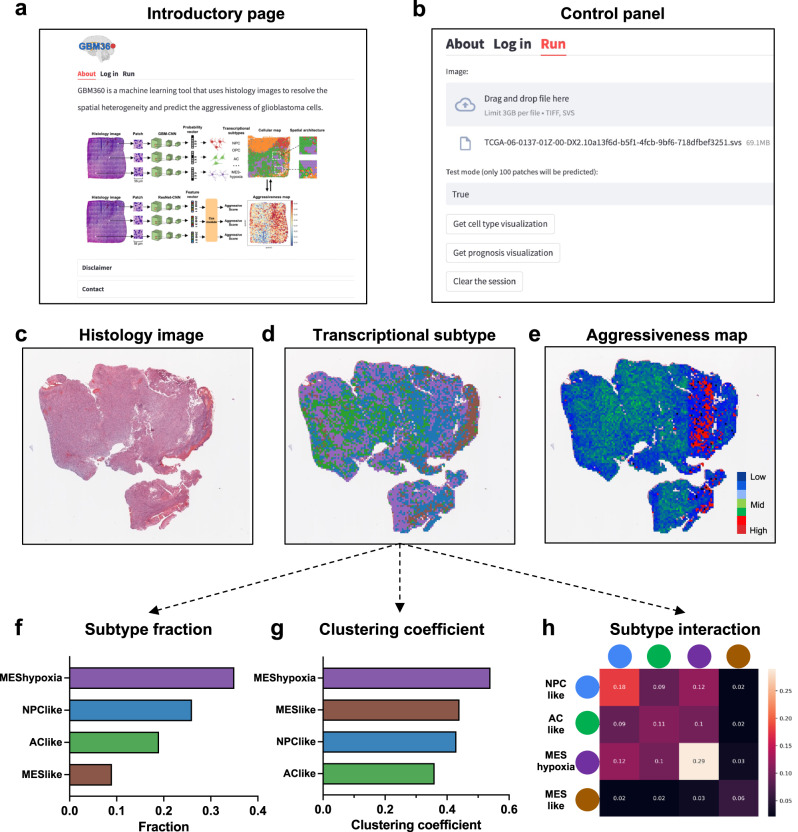
Intra-tumoral heterogeneity and cell-state plasticity are key drivers for the therapeutic resistance of glioblastoma. Here, we investigate the association between spatial cellular organization and glioblastoma prognosis. Leveraging single-cell RNA-seq and spatial transcriptomics data, we develop a deep learning model to predict transcriptional subtypes of glioblastoma cells from histology images. Employing this model, we phenotypically analyze 40 million tissue spots from 410 patients and identify consistent associations between tumor architecture and prognosis across two independent cohorts. Patients with poor prognosis exhibit higher proportions of tumor cells expressing a hypoxia-induced transcriptional program. Furthermore, a clustering pattern of astrocyte-like tumor cells is associated with worse prognosis, while dispersion and connection of the astrocytes with other transcriptional subtypes correlate with decreased risk. To validate these results, we develop a separate deep learning model that utilizes histology images to predict prognosis. Applying this model to spatial transcriptomics data reveal survival-associated regional gene expression programs. Overall, our study presents a scalable approach to unravel the transcriptional heterogeneity of glioblastoma and establishes a critical connection between spatial cellular architecture and clinical outcomes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
