

The cause-effect relation of tuberculosis on incidence of diabetes mellitus
- PMID: 37434784
- PMCID: PMC10330781
- DOI: 10.3389/fcimb.2023.1134036
The cause-effect relation of tuberculosis on incidence of diabetes mellitus
Abstract
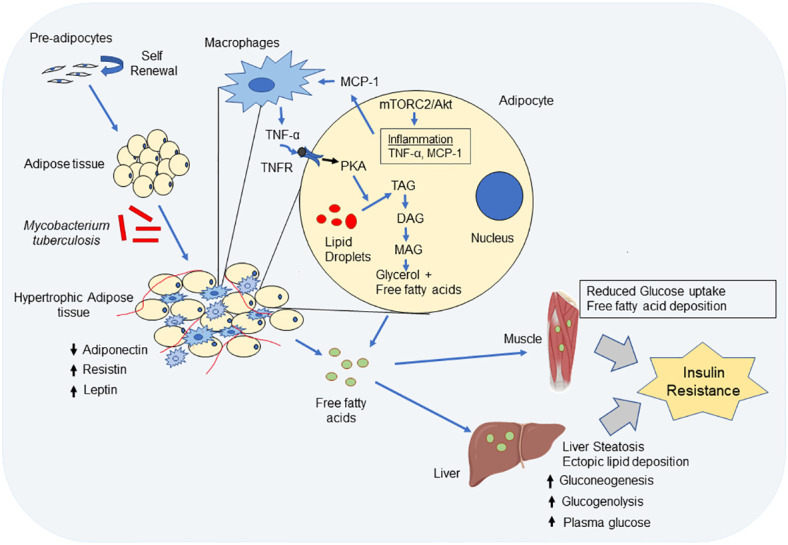
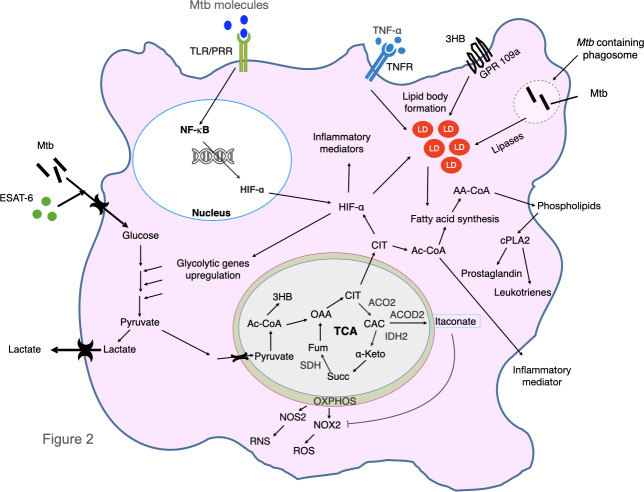
Tuberculosis (TB) is one of the oldest human diseases and is one of the major causes of mortality and morbidity across the Globe. Mycobacterium tuberculosis (Mtb), the causal agent of TB is one of the most successful pathogens known to mankind. Malnutrition, smoking, co-infection with other pathogens like human immunodeficiency virus (HIV), or conditions like diabetes further aggravate the tuberculosis pathogenesis. The association between type 2 diabetes mellitus (DM) and tuberculosis is well known and the immune-metabolic changes during diabetes are known to cause increased susceptibility to tuberculosis. Many epidemiological studies suggest the occurrence of hyperglycemia during active TB leading to impaired glucose tolerance and insulin resistance. However, the mechanisms underlying these effects is not well understood. In this review, we have described possible causal factors like inflammation, host metabolic changes triggered by tuberculosis that could contribute to the development of insulin resistance and type 2 diabetes. We have also discussed therapeutic management of type 2 diabetes during TB, which may help in designing future strategies to cope with TB-DM cases.
Keywords: Mycobacterium tuberculosis; diabetes; inflammation; insulin resistance; therapeutic strategies.
Copyright © 2023 Bisht, Dahiya, Ghosh and Mukhopadhyay.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Impact of diabetes on the natural history of tuberculosis.Diabetes Res Clin Pract. 2014 Nov;106(2):191-9. doi: 10.1016/j.diabres.2014.06.011. Epub 2014 Jul 14. Diabetes Res Clin Pract. 2014. PMID: 25082309 Free PMC article. Review.
-
Stress Hyperglycemia in Patients with Tuberculosis Disease: Epidemiology and Clinical Implications.Curr Diab Rep. 2018 Aug 9;18(9):71. doi: 10.1007/s11892-018-1036-y. Curr Diab Rep. 2018. PMID: 30090969 Free PMC article. Review.
-
Host Metabolic Changes during Mycobacterium Tuberculosis Infection Cause Insulin Resistance in Adult Mice.J Clin Med. 2022 Mar 16;11(6):1646. doi: 10.3390/jcm11061646. J Clin Med. 2022. PMID: 35329973 Free PMC article.
-
Tuberculosis and diabetes mellitus: Relating immune impact of co-morbidity with challenges in disease management in high burden countries.J Clin Tuberc Other Mycobact Dis. 2022 Nov 29;29:100343. doi: 10.1016/j.jctube.2022.100343. eCollection 2022 Dec. J Clin Tuberc Other Mycobact Dis. 2022. PMID: 36478777 Free PMC article.
-
The epidemiology of tuberculosis-associated hyperglycemia in individuals newly screened for type 2 diabetes mellitus: systematic review and meta-analysis.BMC Infect Dis. 2020 Dec 9;20(1):937. doi: 10.1186/s12879-020-05512-7. BMC Infect Dis. 2020. PMID: 33297969 Free PMC article.
Cited by
-
Metabolic insights into HIV/TB co-infection: an untargeted urinary metabolomics approach.Metabolomics. 2024 Jul 16;20(4):78. doi: 10.1007/s11306-024-02148-5. Metabolomics. 2024. PMID: 39014031 Free PMC article.
-
Host-directed therapy against mycobacterium tuberculosis infections with diabetes mellitus.Front Immunol. 2024 Jan 8;14:1305325. doi: 10.3389/fimmu.2023.1305325. eCollection 2023. Front Immunol. 2024. PMID: 38259491 Free PMC article. Review.
-
Examining the efficacy of treatment outcomes for patients with pulmonary tuberculosis in Western China: A retrospective study in a region of high incidence.BMC Public Health. 2025 Apr 11;25(1):1360. doi: 10.1186/s12889-025-22543-4. BMC Public Health. 2025. PMID: 40217246 Free PMC article.
-
Associations between type 1 diabetes and pulmonary tuberculosis: a bidirectional mendelian randomization study.Diabetol Metab Syndr. 2024 Mar 5;16(1):60. doi: 10.1186/s13098-024-01296-x. Diabetol Metab Syndr. 2024. PMID: 38443967 Free PMC article.
-
Unrecognized Tuberculosis: Risk Factors for Smear-Positive/Cavitary Asymptomatic Cases.Open Forum Infect Dis. 2025 Mar 22;12(4):ofaf176. doi: 10.1093/ofid/ofaf176. eCollection 2025 Apr. Open Forum Infect Dis. 2025. PMID: 40242060 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical