

Duchenne muscular dystrophy: disease mechanism and therapeutic strategies
- PMID: 37435300
- PMCID: PMC10330733
- DOI: 10.3389/fphys.2023.1183101
Duchenne muscular dystrophy: disease mechanism and therapeutic strategies
Abstract
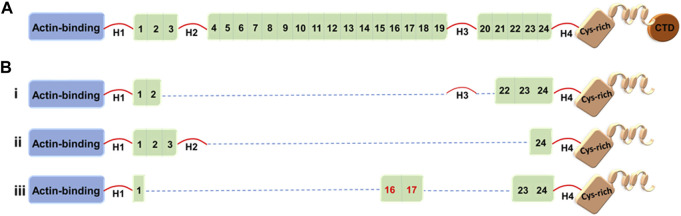
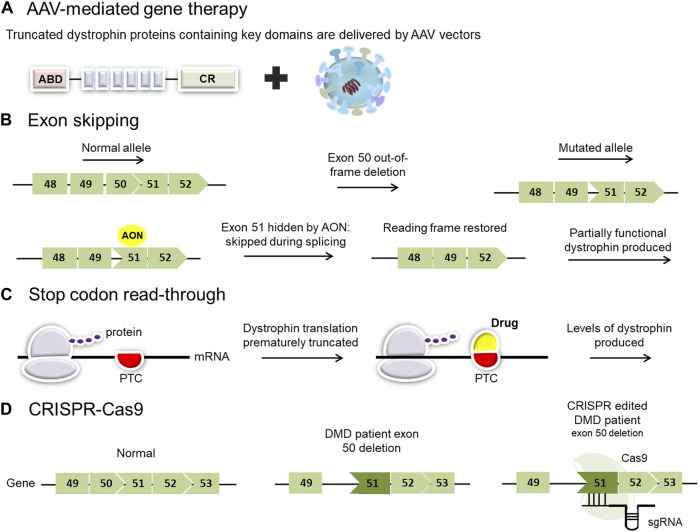
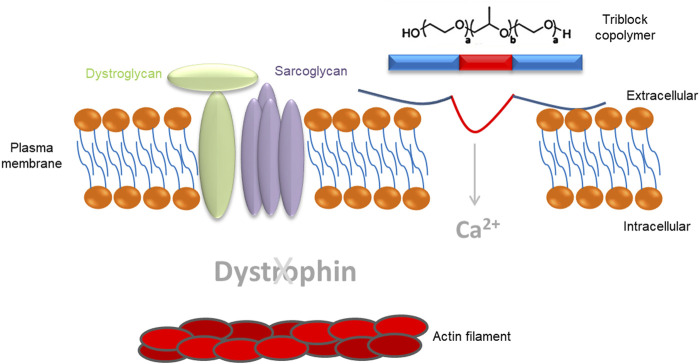
Duchenne muscular dystrophy (DMD) is a severe, progressive, and ultimately fatal disease of skeletal muscle wasting, respiratory insufficiency, and cardiomyopathy. The identification of the dystrophin gene as central to DMD pathogenesis has led to the understanding of the muscle membrane and the proteins involved in membrane stability as the focal point of the disease. The lessons learned from decades of research in human genetics, biochemistry, and physiology have culminated in establishing the myriad functionalities of dystrophin in striated muscle biology. Here, we review the pathophysiological basis of DMD and discuss recent progress toward the development of therapeutic strategies for DMD that are currently close to or are in human clinical trials. The first section of the review focuses on DMD and the mechanisms contributing to membrane instability, inflammation, and fibrosis. The second section discusses therapeutic strategies currently used to treat DMD. This includes a focus on outlining the strengths and limitations of approaches directed at correcting the genetic defect through dystrophin gene replacement, modification, repair, and/or a range of dystrophin-independent approaches. The final section highlights the different therapeutic strategies for DMD currently in clinical trials.
Keywords: Duchenne muscular dystrophy; dystrophin; muscle disease; pathophysiology; skeletal muscle; therapeutic strategies.
Copyright © 2023 Bez Batti Angulski, Hosny, Cohen, Martin, Hahn, Bauer and Metzger.
Conflict of interest statement
JM is on the scientific advisory board of and holds shares in Phrixus Pharmaceuticals Inc., a company developing novel therapeutics for heart failure. The terms of this arrangement have been reviewed and approved by the University of Minnesota in accordance with its conflict-of-interest policies.
Figures
References
-
- Aartsma‐Rus A., Van Deutekom J. C. T., Fokkema I. F., Van Ommen G. B., Den Dunnen J. T. (2006). Entries in the leiden duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading‐frame rule. Muscle and Nerve Official J. Am. Assoc. Electrodiagn. Med. 34, 135–144. 10.1002/mus.20586 - DOI - PubMed
-
- Abmayr S., Chamberlain J. (2006). The structure and function of dystrophin. Molecular mechanisms of muscular dystrophies. Georget. Landes Biosci., 14–34.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources