

Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform
- PMID: 37438352
- PMCID: PMC10338508
- DOI: 10.1038/s41467-023-39869-5
Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform
Abstract
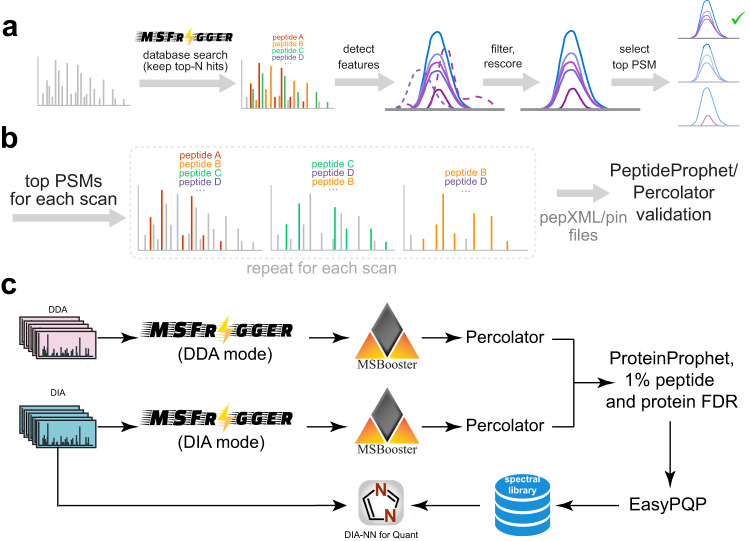
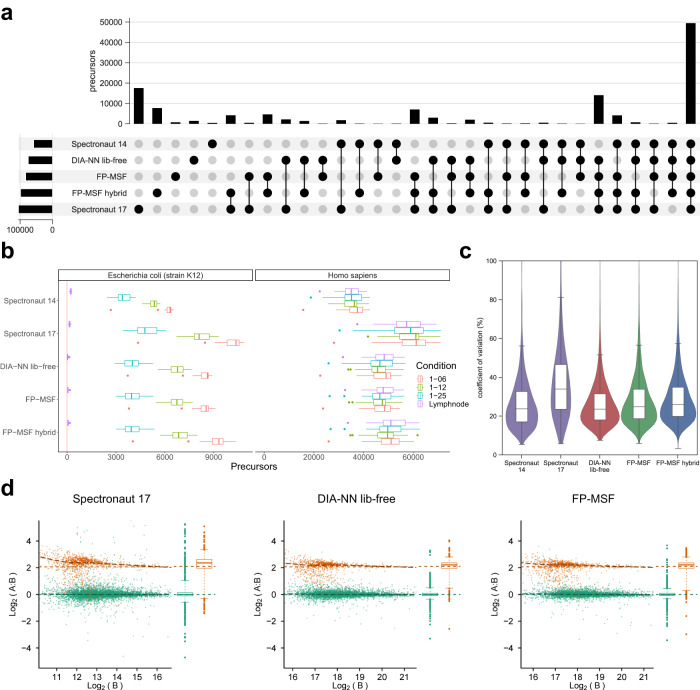
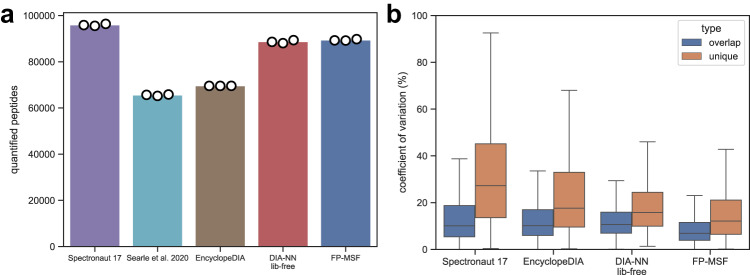
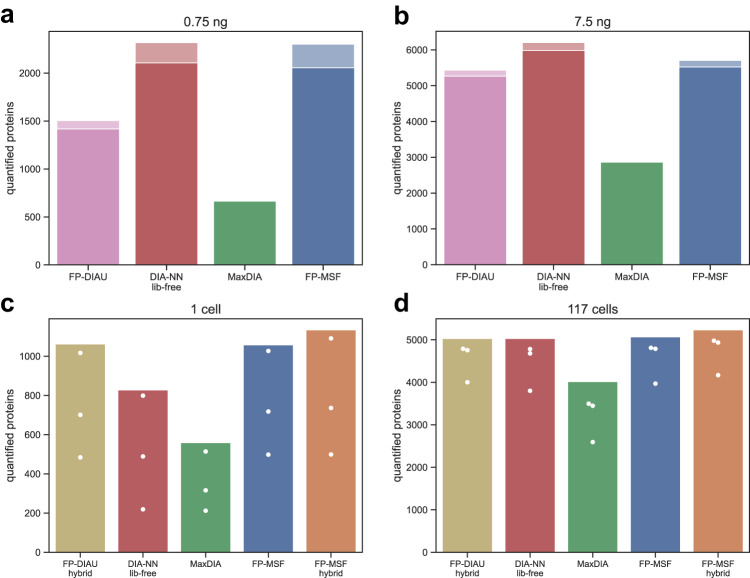
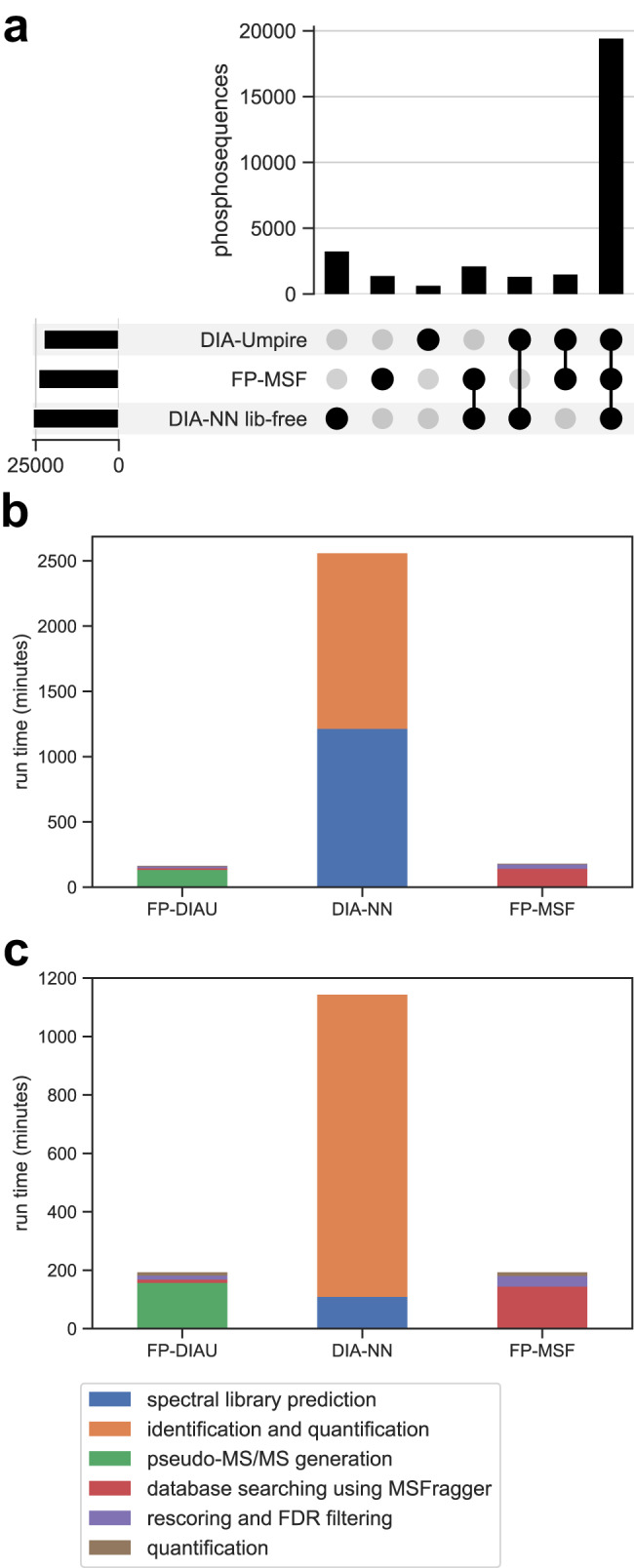
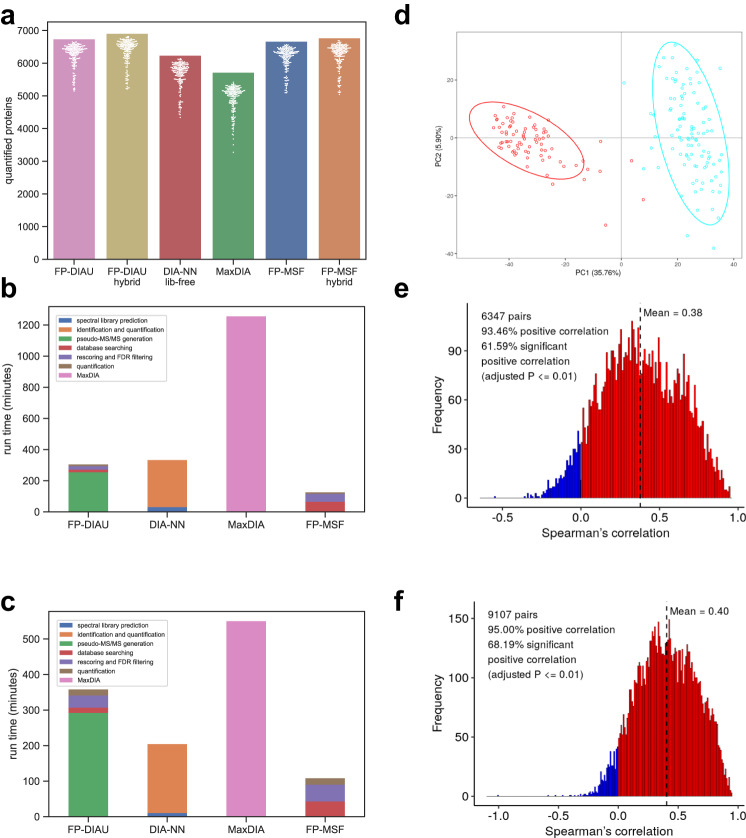
Liquid chromatography (LC) coupled with data-independent acquisition (DIA) mass spectrometry (MS) has been increasingly used in quantitative proteomics studies. Here, we present a fast and sensitive approach for direct peptide identification from DIA data, MSFragger-DIA, which leverages the unmatched speed of the fragment ion indexing-based search engine MSFragger. Different from most existing methods, MSFragger-DIA conducts a database search of the DIA tandem mass (MS/MS) spectra prior to spectral feature detection and peak tracing across the LC dimension. To streamline the analysis of DIA data and enable easy reproducibility, we integrate MSFragger-DIA into the FragPipe computational platform for seamless support of peptide identification and spectral library building from DIA, data-dependent acquisition (DDA), or both data types combined. We compare MSFragger-DIA with other DIA tools, such as DIA-Umpire based workflow in FragPipe, Spectronaut, DIA-NN library-free, and MaxDIA. We demonstrate the fast, sensitive, and accurate performance of MSFragger-DIA across a variety of sample types and data acquisition schemes, including single-cell proteomics, phosphoproteomics, and large-scale tumor proteome profiling studies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kitata, R. B., Yang, J. C. & Chen, Y. J. Advances in data-independent acquisition mass spectrometry towards comprehensive digital proteome landscape. Mass Spectrom. Rev. e21781 (2022). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
