

SEESAW: detecting isoform-level allelic imbalance accounting for inferential uncertainty
- PMID: 37438847
- PMCID: PMC10337143
- DOI: 10.1186/s13059-023-03003-x
SEESAW: detecting isoform-level allelic imbalance accounting for inferential uncertainty
Abstract
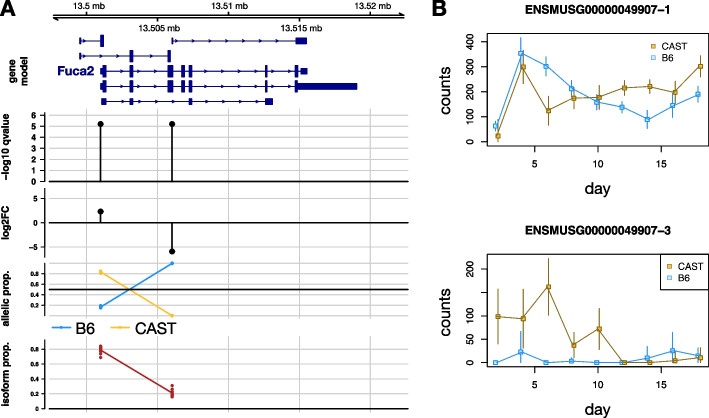
Detecting allelic imbalance at the isoform level requires accounting for inferential uncertainty, caused by multi-mapping of RNA-seq reads. Our proposed method, SEESAW, uses Salmon and Swish to offer analysis at various levels of resolution, including gene, isoform, and aggregating isoforms to groups by transcription start site. The aggregation strategies strengthen the signal for transcripts with high uncertainty. The SEESAW suite of methods is shown to have higher power than other allelic imbalance methods when there is isoform-level allelic imbalance. We also introduce a new test for detecting imbalance that varies across a covariate, such as time.
© 2023. The Author(s).
Conflict of interest statement
R.P. is a co-founder of Ocean Genomics Inc.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
