

Likelihood-Based Tests of Species Tree Hypotheses
- PMID: 37440530
- PMCID: PMC10368450
- DOI: 10.1093/molbev/msad159
Likelihood-Based Tests of Species Tree Hypotheses
Abstract
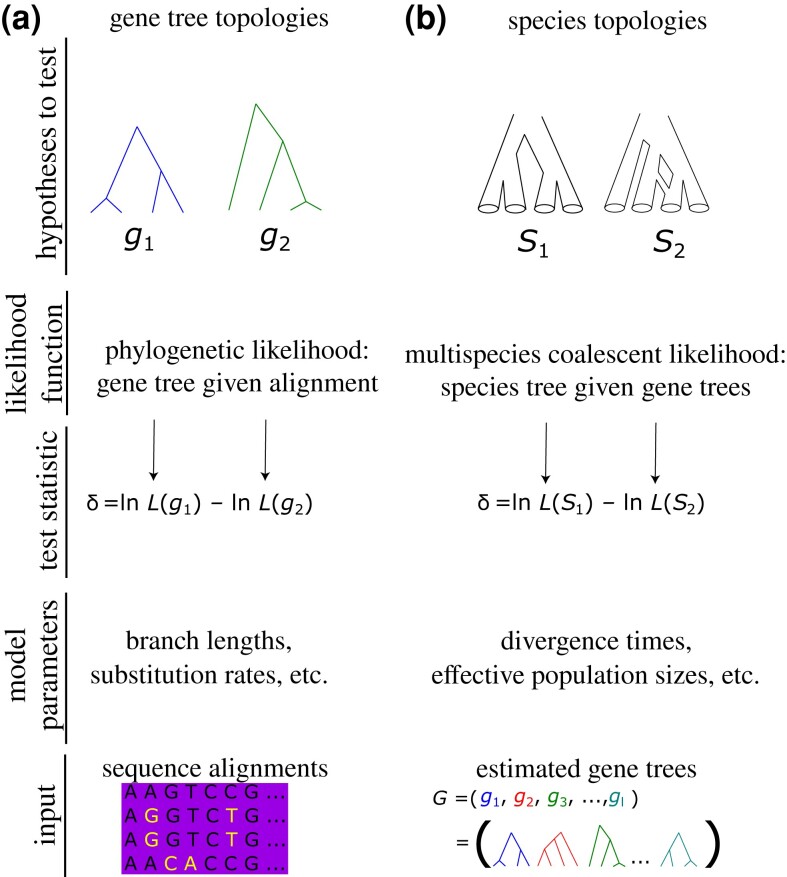
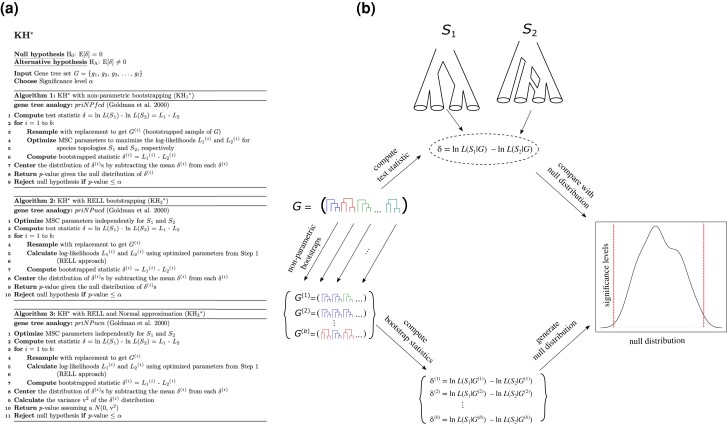
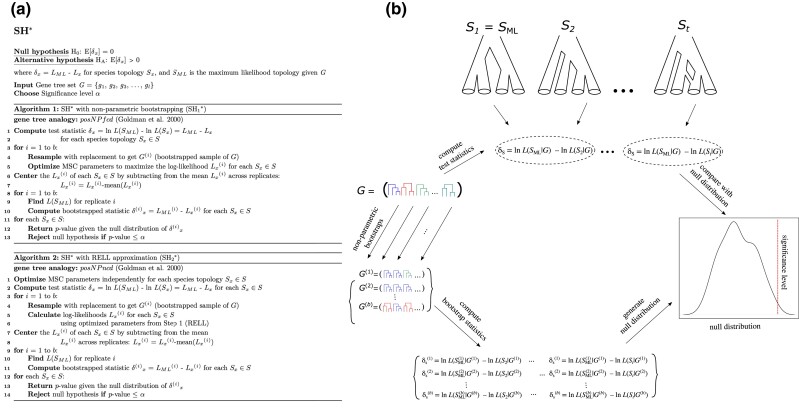
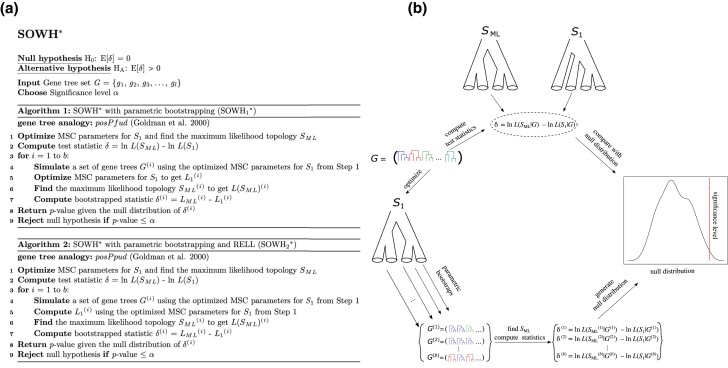
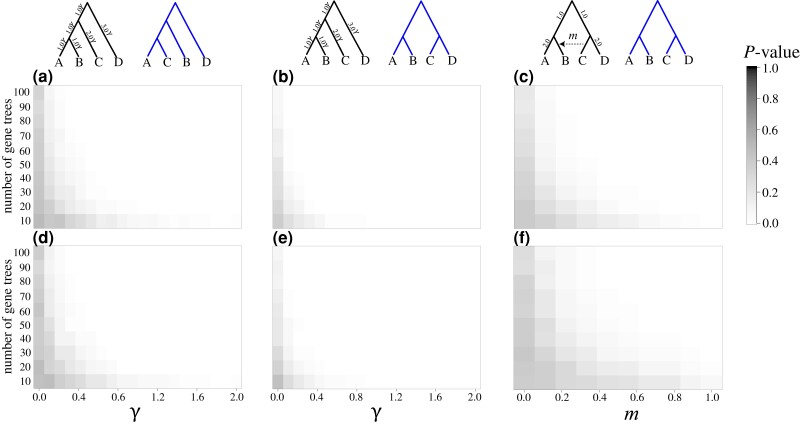
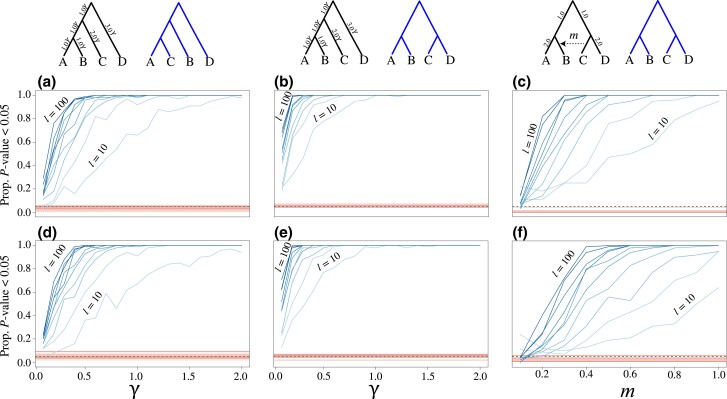
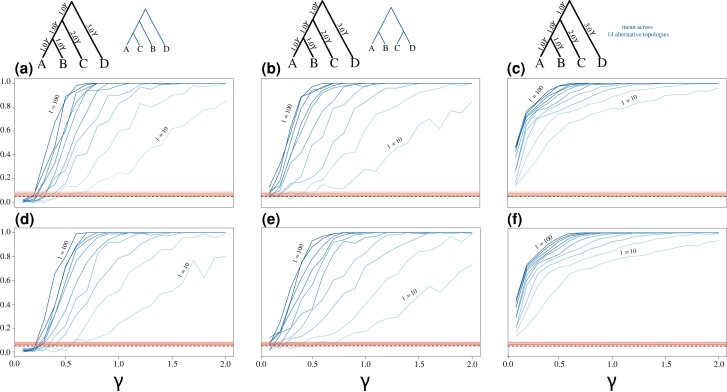
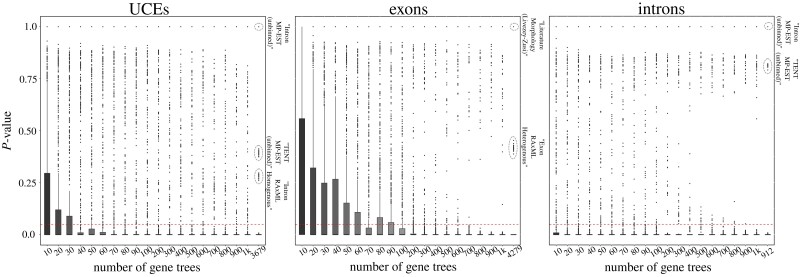
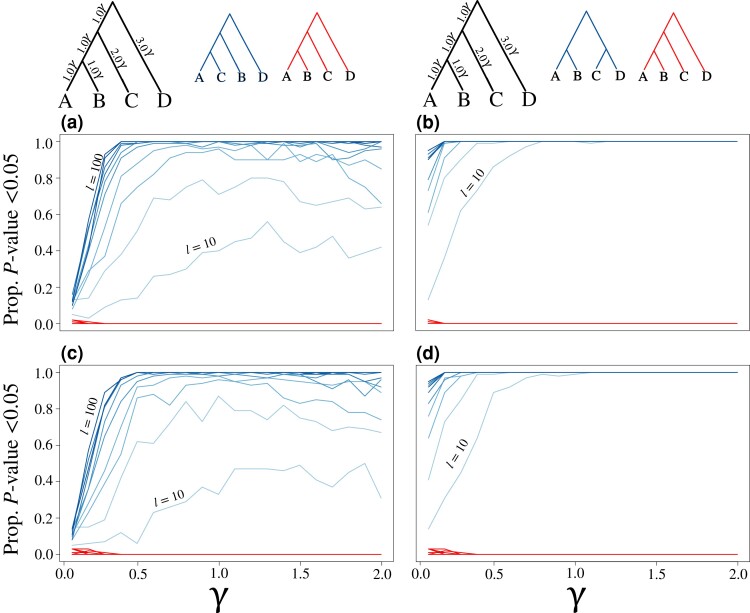
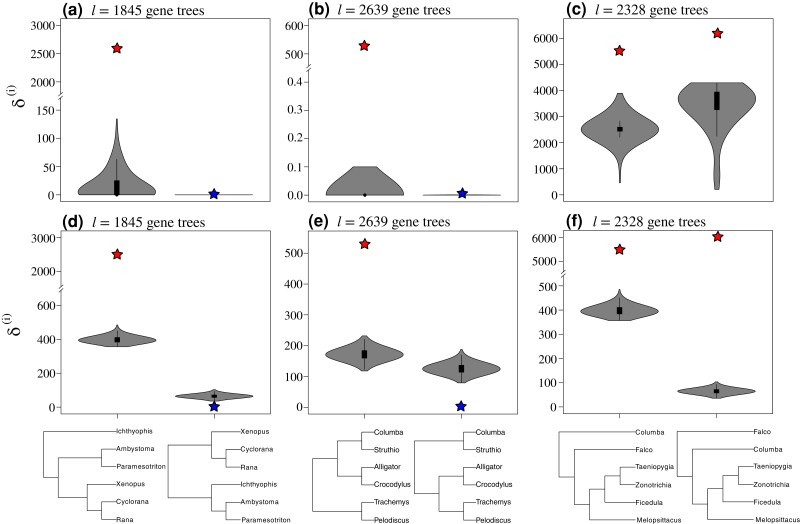
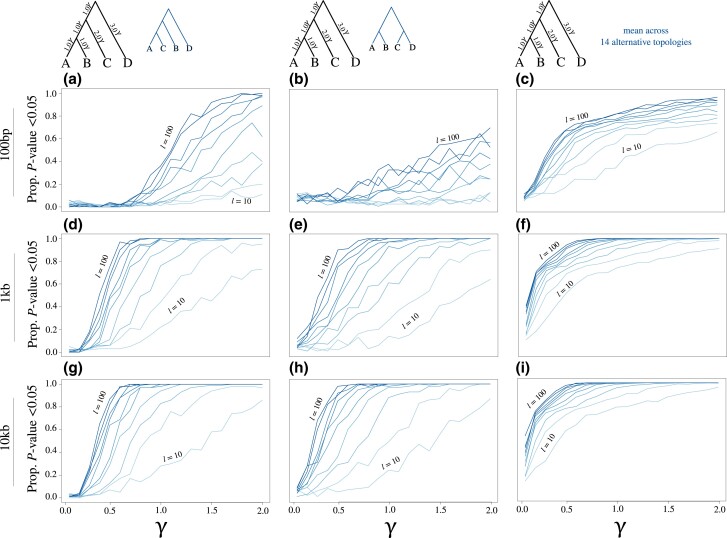
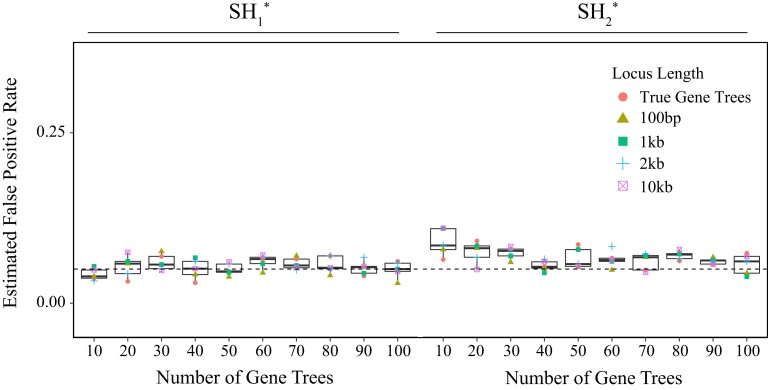
Likelihood-based tests of phylogenetic trees are a foundation of modern systematics. Over the past decade, an enormous wealth and diversity of model-based approaches have been developed for phylogenetic inference of both gene trees and species trees. However, while many techniques exist for conducting formal likelihood-based tests of gene trees, such frameworks are comparatively underdeveloped and underutilized for testing species tree hypotheses. To date, widely used tests of tree topology are designed to assess the fit of classical models of molecular sequence data and individual gene trees and thus are not readily applicable to the problem of species tree inference. To address this issue, we derive several analogous likelihood-based approaches for testing topologies using modern species tree models and heuristic algorithms that use gene tree topologies as input for maximum likelihood estimation under the multispecies coalescent. For the purpose of comparing support for species trees, these tests leverage the statistical procedures of their original gene tree-based counterparts that have an extended history for testing phylogenetic hypotheses at a single locus. We discuss and demonstrate a number of applications, limitations, and important considerations of these tests using simulated and empirical phylogenomic data sets that include both bifurcating topologies and reticulate network models of species relationships. Finally, we introduce the open-source R package SpeciesTopoTestR (SpeciesTopology Tests in R) that includes a suite of functions for conducting formal likelihood-based tests of species topologies given a set of input gene tree topologies.
Keywords: bootstrap; maximum likelihood; multispecies coalescent; phylogenetic networks; phylogenomics.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
Similar articles
-
Modern Phylogenomics: Building Phylogenetic Trees Using the Multispecies Coalescent Model.Methods Mol Biol. 2019;1910:211-239. doi: 10.1007/978-1-4939-9074-0_7. Methods Mol Biol. 2019. PMID: 31278666
-
Coalescent-based species tree inference from gene tree topologies under incomplete lineage sorting by maximum likelihood.Evolution. 2012 Mar;66(3):763-775. doi: 10.1111/j.1558-5646.2011.01476.x. Epub 2011 Nov 2. Evolution. 2012. PMID: 22380439
-
Challenges in Species Tree Estimation Under the Multispecies Coalescent Model.Genetics. 2016 Dec;204(4):1353-1368. doi: 10.1534/genetics.116.190173. Genetics. 2016. PMID: 27927902 Free PMC article. Review.
-
STELLS2: fast and accurate coalescent-based maximum likelihood inference of species trees from gene tree topologies.Bioinformatics. 2017 Jun 15;33(12):1789-1797. doi: 10.1093/bioinformatics/btx079. Bioinformatics. 2017. PMID: 28186220
-
Coalescent methods for estimating phylogenetic trees.Mol Phylogenet Evol. 2009 Oct;53(1):320-8. doi: 10.1016/j.ympev.2009.05.033. Epub 2009 Jun 6. Mol Phylogenet Evol. 2009. PMID: 19501178 Review.
Cited by
-
Genome-wide identification and mining elite allele variation of the Monoacylglycerol lipase (MAGL) gene family in upland cotton (Gossypium hirsutum L.).BMC Plant Biol. 2024 Jun 21;24(1):587. doi: 10.1186/s12870-024-05297-w. BMC Plant Biol. 2024. PMID: 38902638 Free PMC article.
-
Mass development of a filamentous and likely nitrophilous aerophytic green alga on tree bark: Apatococcus ammoniophilus sp. nov. (Chlorophyta, Trebouxiophyceae).Front Microbiol. 2025 Jul 23;16:1633308. doi: 10.3389/fmicb.2025.1633308. eCollection 2025. Front Microbiol. 2025. PMID: 40771691 Free PMC article.
References
-
- Adams RH, Castoe TA. 2019. Statistical binning leads to profound model violation due to gene tree error incurred by trying to avoid gene tree error. Mol Phyl Evol. 134:164–171. - PubMed
-
- Adams RH, Schield DR, Card DC, Castoe TA. 2018. Assessing the impacts of positive selection on coalescent-based species tree estimation and species delimitation. Syst Biol. 67:1076–1090. - PubMed
-
- Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 55:539–552. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
