

Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases
- PMID: 37443254
- PMCID: PMC10836580
- DOI: 10.1038/s41588-023-01443-6
Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases
Abstract
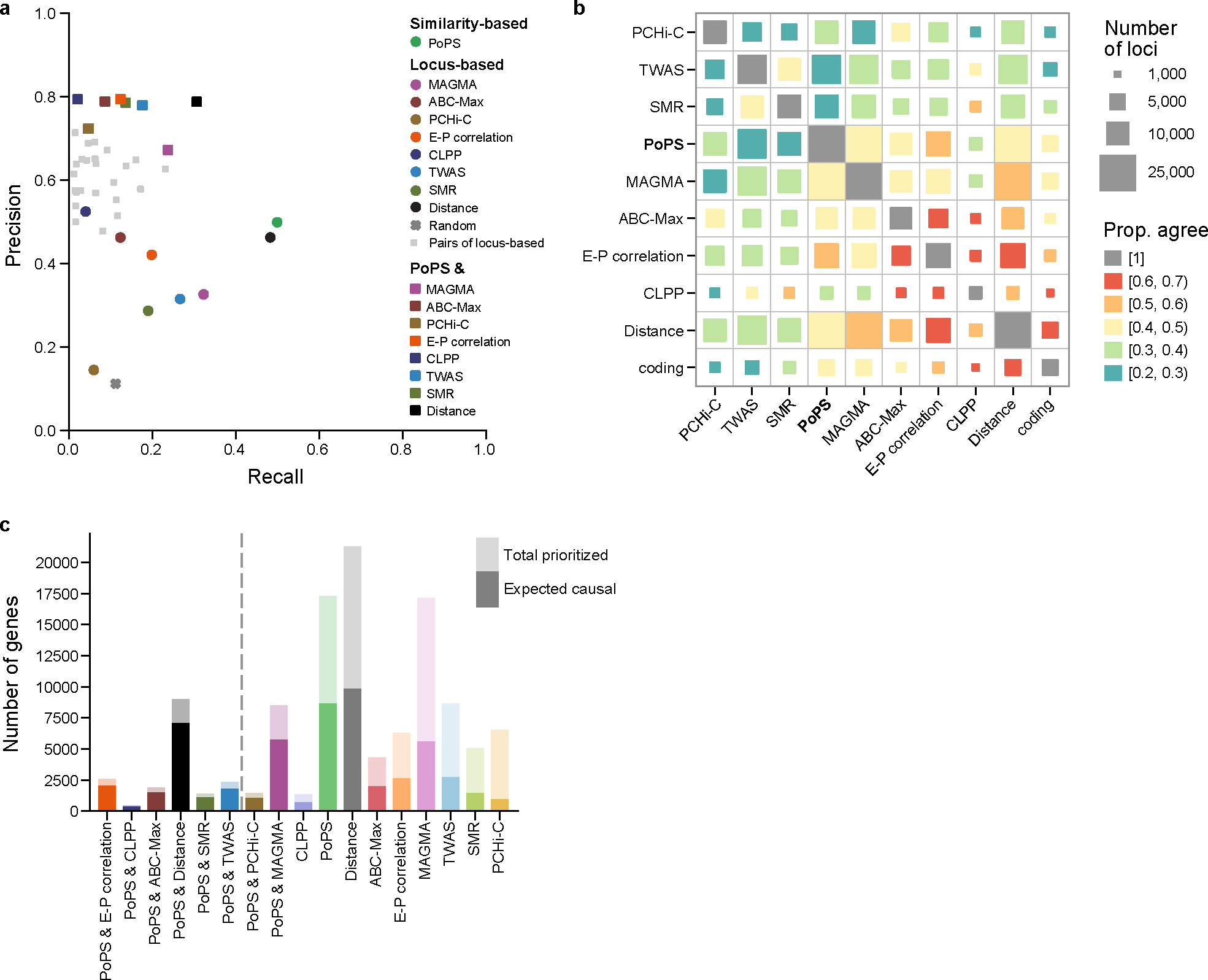
Genome-wide association studies (GWASs) are a valuable tool for understanding the biology of complex human traits and diseases, but associated variants rarely point directly to causal genes. In the present study, we introduce a new method, polygenic priority score (PoPS), that learns trait-relevant gene features, such as cell-type-specific expression, to prioritize genes at GWAS loci. Using a large evaluation set of genes with fine-mapped coding variants, we show that PoPS and the closest gene individually outperform other gene prioritization methods, but observe the best overall performance by combining PoPS with orthogonal methods. Using this combined approach, we prioritize 10,642 unique gene-trait pairs across 113 complex traits and diseases with high precision, finding not only well-established gene-trait relationships but nominating new genes at unresolved loci, such as LGR4 for estimated glomerular filtration rate and CCR7 for deep vein thrombosis. Overall, we demonstrate that PoPS provides a powerful addition to the gene prioritization toolbox.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests
J.C.U reports compensation from consulting services with Goldfinch Bio and is an employee of Illumina. R.S.F. is an employee of Vertex Pharmaceuticals Incorporated. C.P.F. is an employee of Bristol Myers Squibb. J.O.M. reports compensation for consulting services with Cellarity. A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov until July 31, 2020. From August 1, 2020, A.R. is an employee of Genentech. J.N.H. served on the Scientific Advisory Board of and consults for Camp4 Therapeutics. E.S.L. serves on the Board of Directors for Codiak BioSciences and Neon Therapeutics, and serves on the Scientific Advisory Board of F-Prime Capital Partners and Third Rock Ventures; he is also affiliated with several non-profit organizations including serving on the Board of Directors of the Innocence Project, Count Me In, and Biden Cancer Initiative, and the Board of Trustees for the Parker Institute for Cancer Immunotherapy. He has served and continues to serve on various federal advisory committees. The remaining authors declare no competing interests.
Figures















References
METHODS-ONLY REFERENCES
-
- Baglama J & Reichel L Restarted block Lanczos bidiagonalization methods. Numer. Algorithms 43, 251–272 (2007).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
