

α-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure
- PMID: 37443257
- PMCID: PMC10474270
- DOI: 10.1038/s41422-023-00844-w
α-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure
Abstract
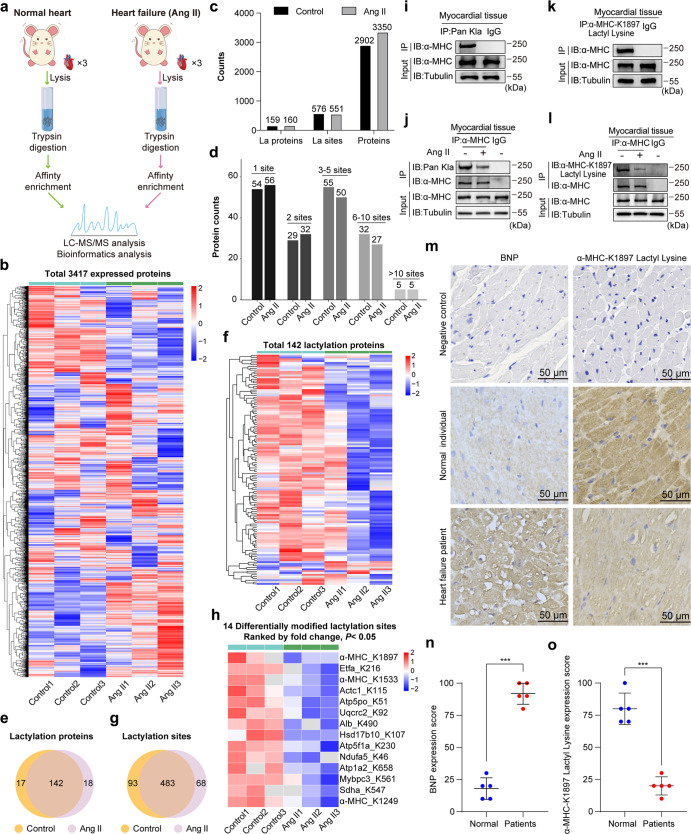
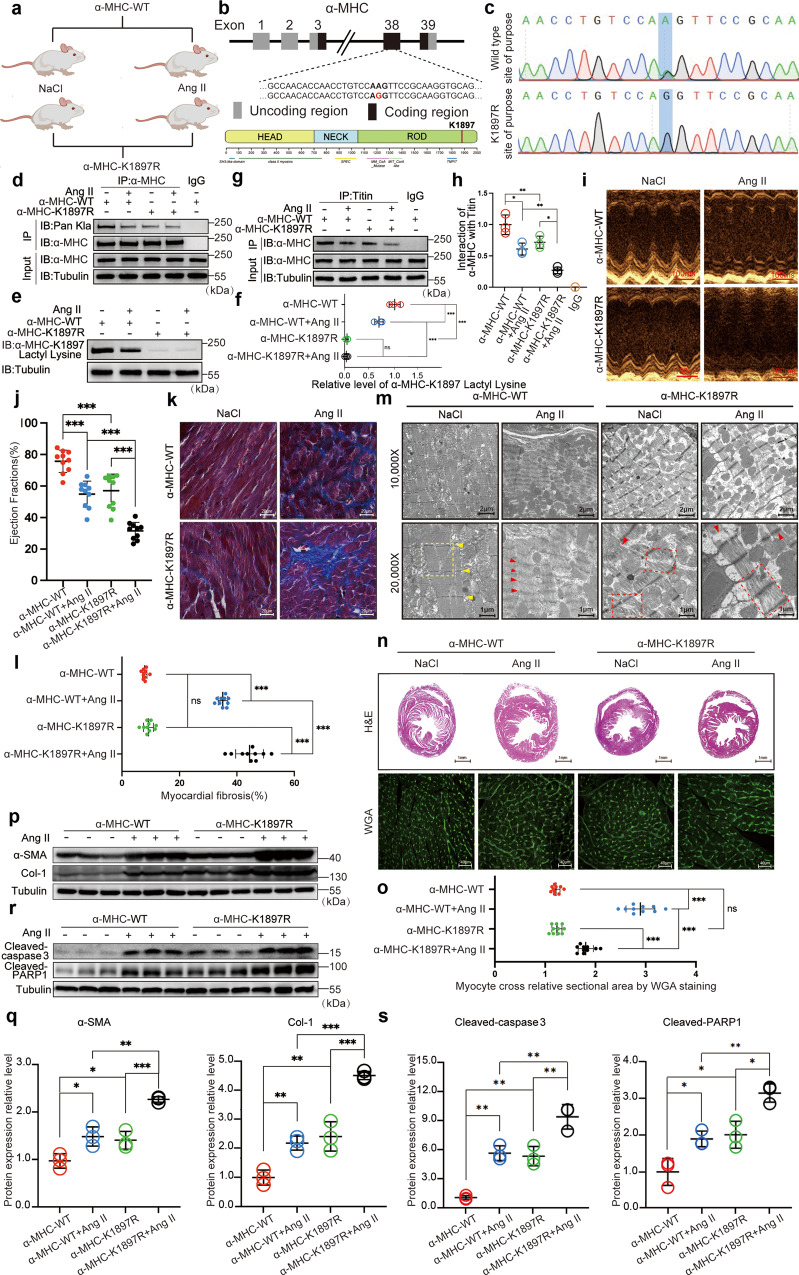
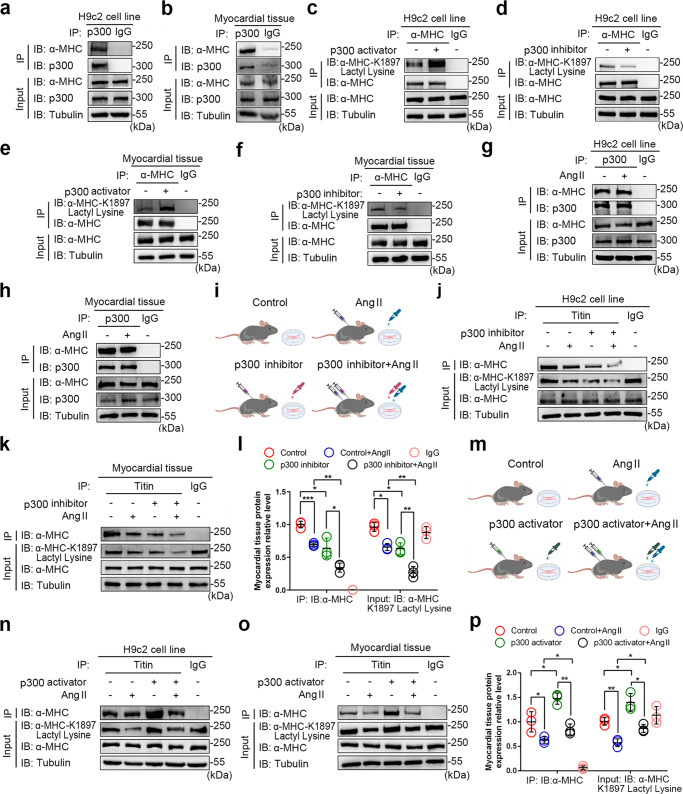
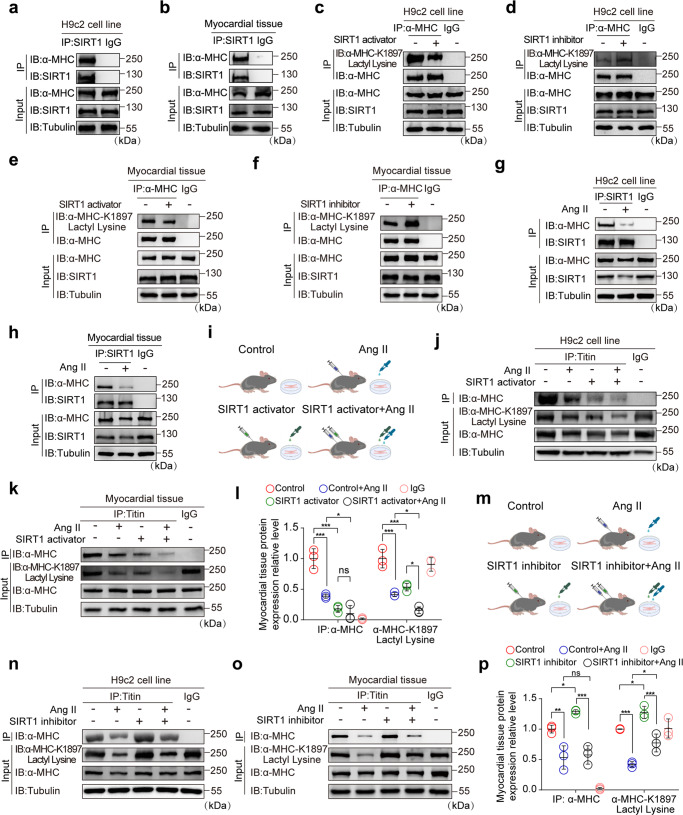
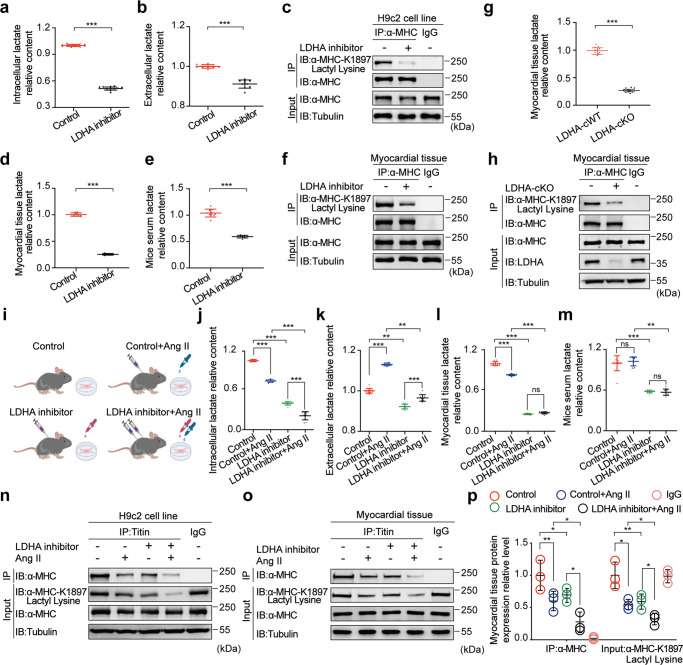
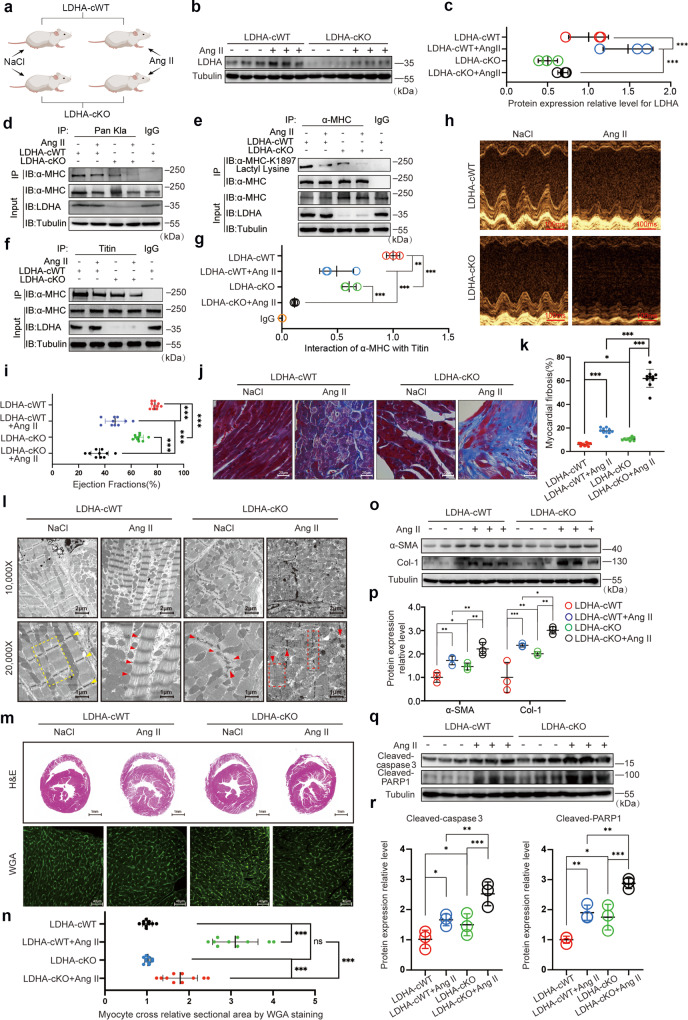
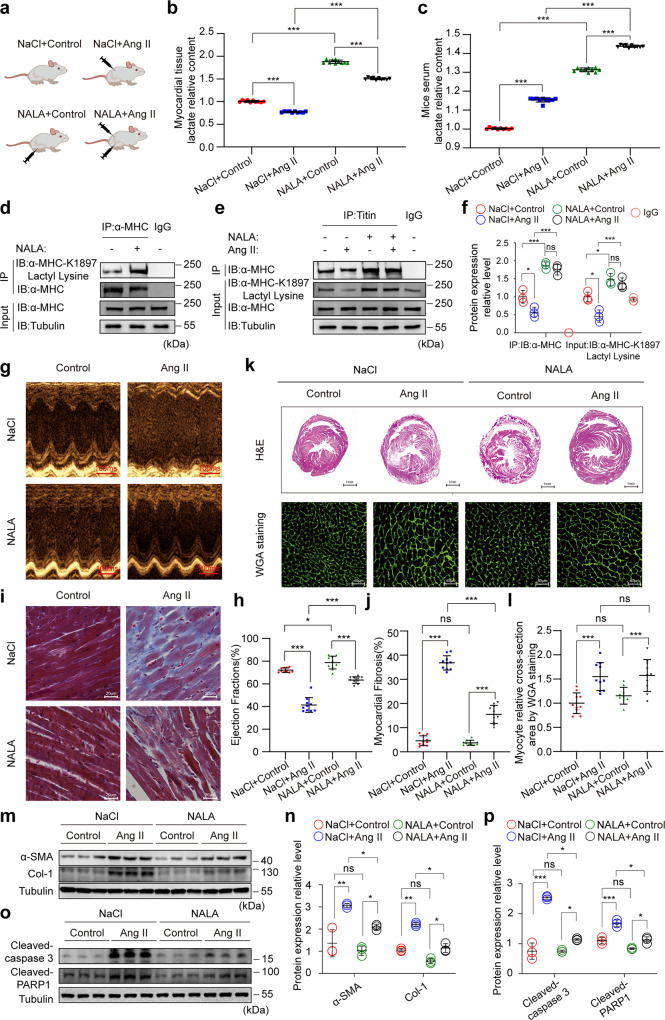
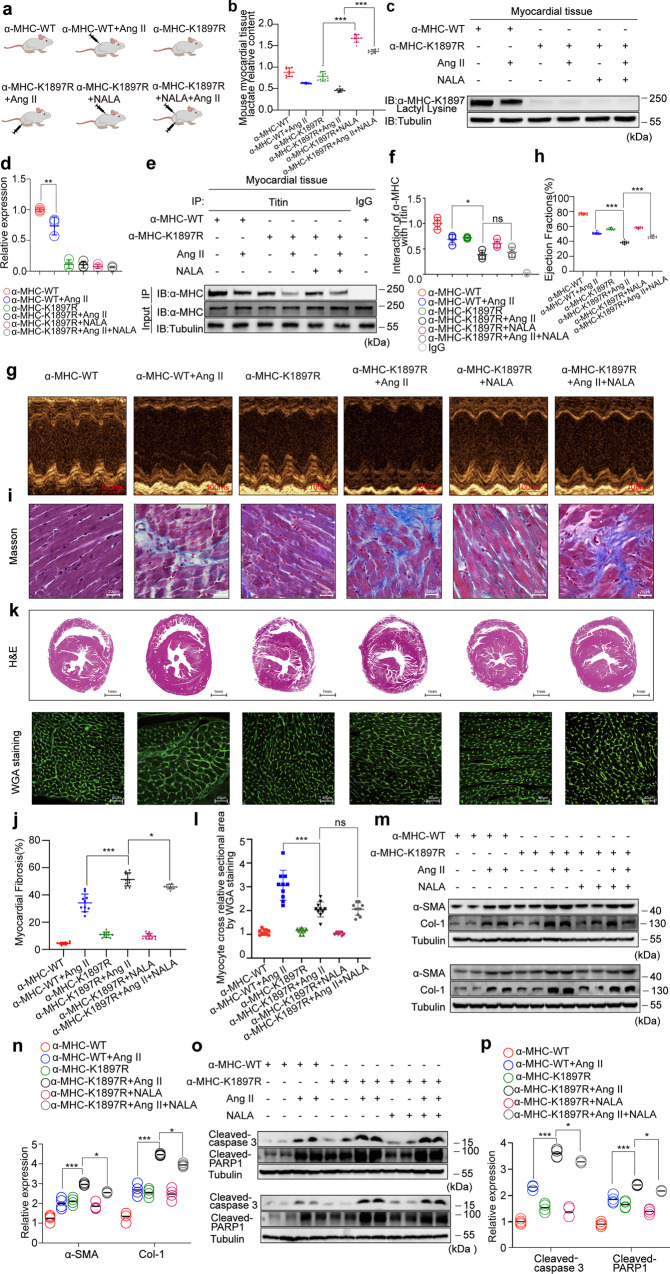
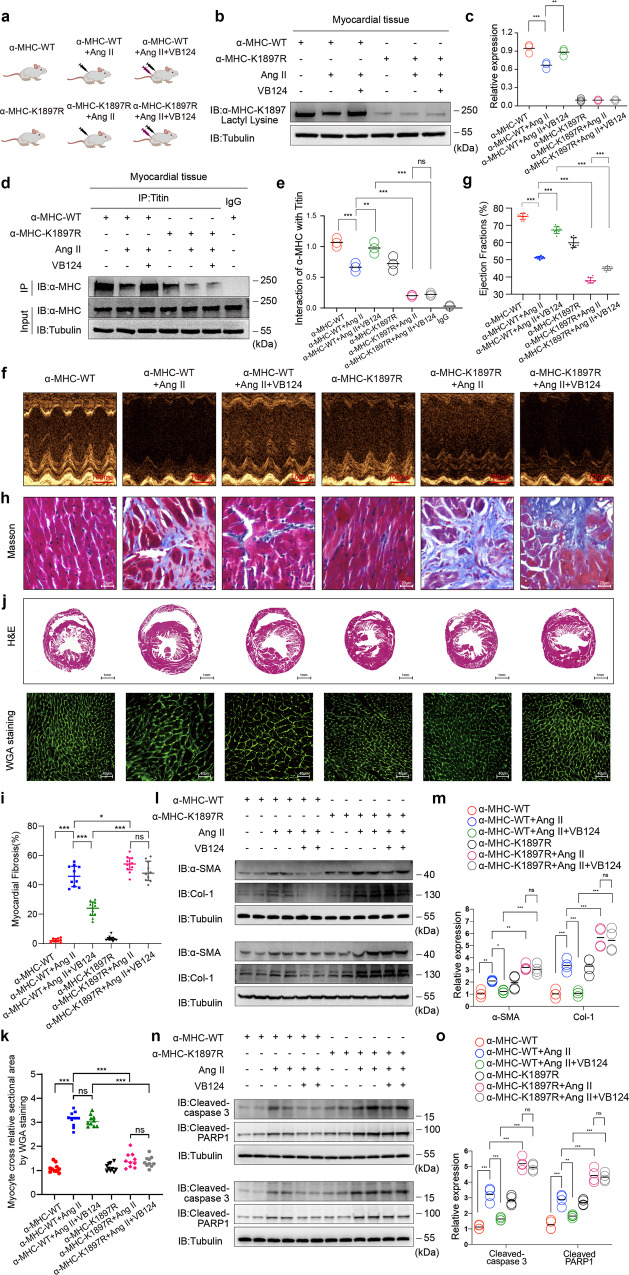
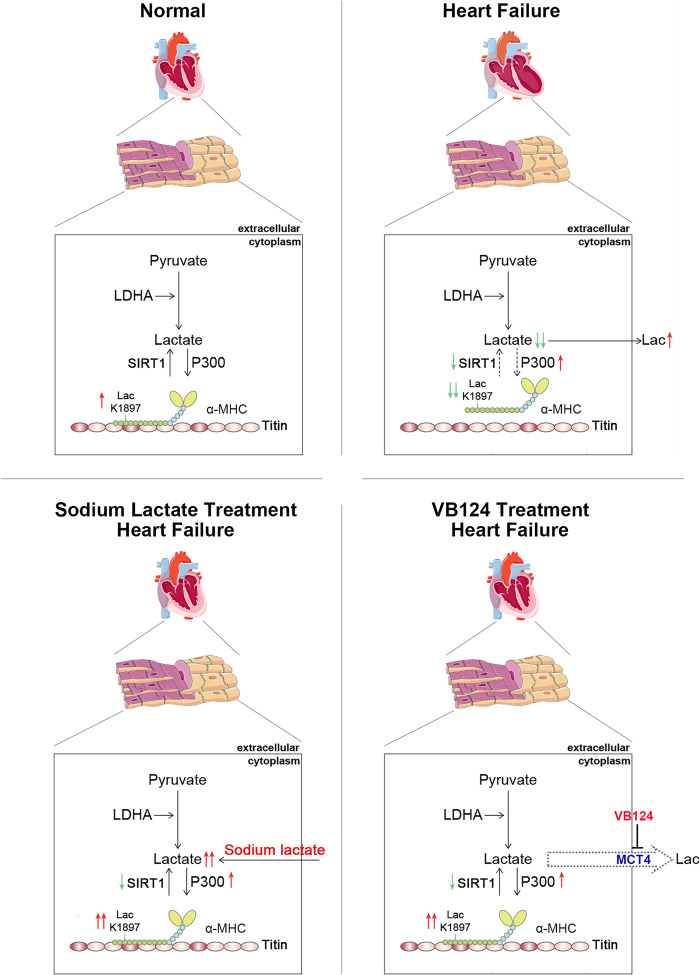
The sarcomeric interaction of α-myosin heavy chain (α-MHC) with Titin is vital for cardiac structure and contraction. However, the mechanism regulating this interaction in normal and failing hearts remains unknown. Lactate is a crucial energy substrate of the heart. Here, we identify that α-MHC undergoes lactylation on lysine 1897 to regulate the interaction of α-MHC with Titin. We observed a reduction of α-MHC K1897 lactylation in mice and patients with heart failure. Loss of K1897 lactylation in α-MHC K1897R knock-in mice reduces α-MHC-Titin interaction and leads to impaired cardiac structure and function. Furthermore, we identified that p300 and Sirtuin 1 act as the acyltransferase and delactylase of α-MHC, respectively. Decreasing lactate production by chemical or genetic manipulation reduces α-MHC lactylation, impairs α-MHC-Titin interaction and worsens heart failure. By contrast, upregulation of the lactate concentration by administering sodium lactate or inhibiting the pivotal lactate transporter in cardiomyocytes can promote α-MHC K1897 lactylation and α-MHC-Titin interaction, thereby alleviating heart failure. In conclusion, α-MHC lactylation is dynamically regulated and an important determinant of overall cardiac structure and function. Excessive lactate efflux and consumption by cardiomyocytes may decrease the intracellular lactate level, which is the main cause of reduced α-MHC K1897 lactylation during myocardial injury. Our study reveals that cardiac metabolism directly modulates the sarcomeric structure and function through lactate-dependent modification of α-MHC.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Lactylation regulates cardiac function.Cell Res. 2023 Sep;33(9):653-654. doi: 10.1038/s41422-023-00857-5. Cell Res. 2023. PMID: 37488305 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
