

Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision
- PMID: 37443337
- PMCID: PMC10870000
- DOI: 10.1038/s41592-023-01944-6
Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision
Erratum in
-
Author Correction: Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision.Nat Methods. 2025 Feb;22(2):448. doi: 10.1038/s41592-024-02569-z. Nat Methods. 2025. PMID: 39825083 No abstract available.
Abstract
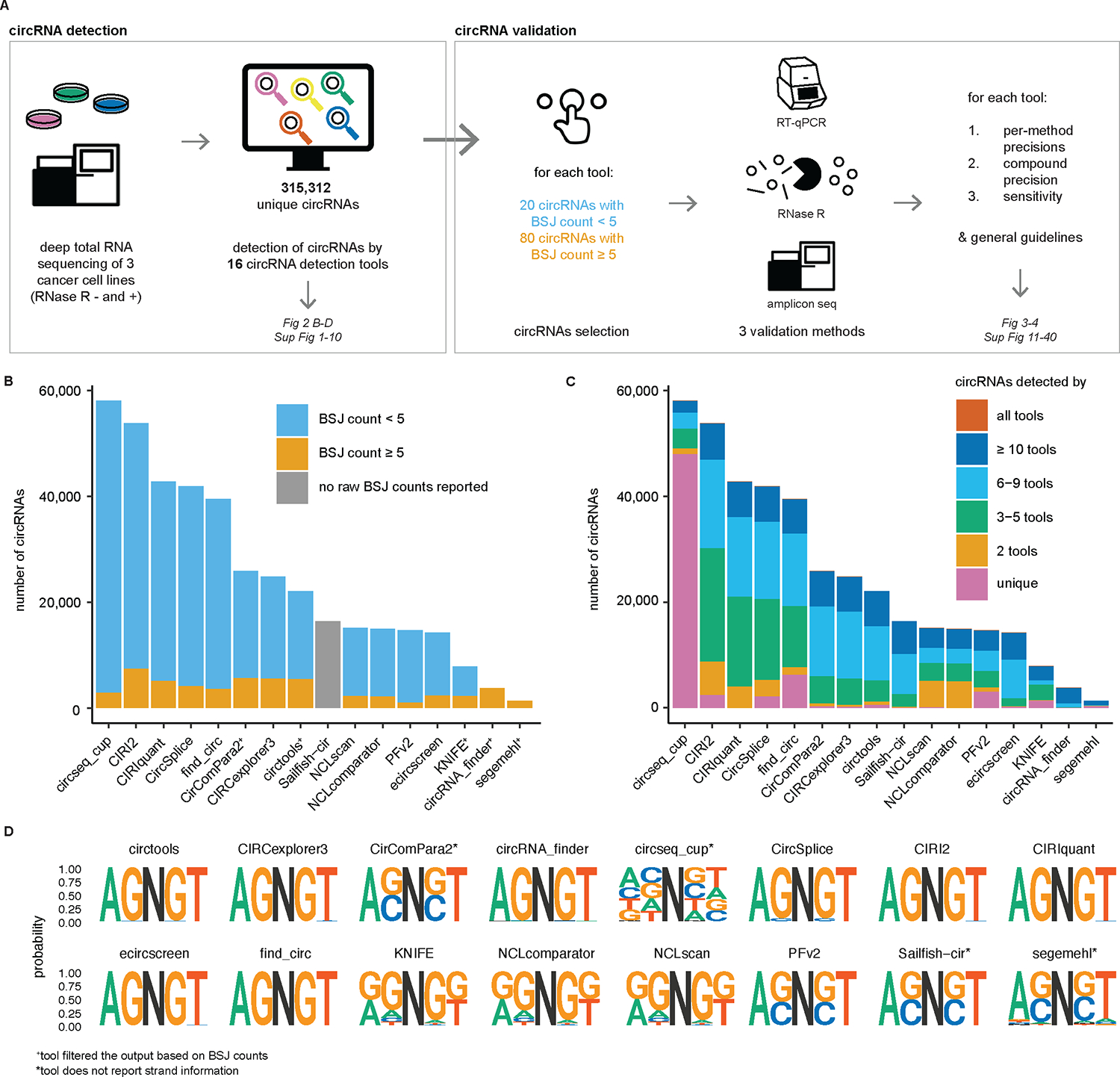
The detection of circular RNA molecules (circRNAs) is typically based on short-read RNA sequencing data processed using computational tools. Numerous such tools have been developed, but a systematic comparison with orthogonal validation is missing. Here, we set up a circRNA detection tool benchmarking study, in which 16 tools detected more than 315,000 unique circRNAs in three deeply sequenced human cell types. Next, 1,516 predicted circRNAs were validated using three orthogonal methods. Generally, tool-specific precision is high and similar (median of 98.8%, 96.3% and 95.5% for qPCR, RNase R and amplicon sequencing, respectively) whereas the sensitivity and number of predicted circRNAs (ranging from 1,372 to 58,032) are the most significant differentiators. Of note, precision values are lower when evaluating low-abundance circRNAs. We also show that the tools can be used complementarily to increase detection sensitivity. Finally, we offer recommendations for future circRNA detection and validation.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures



Comment in
-
Circular RNA detection pipelines yield divergent sets of circular RNAs.Nat Methods. 2023 Aug;20(8):1135-1136. doi: 10.1038/s41592-023-01945-5. Nat Methods. 2023. PMID: 37443339 No abstract available.
References
-
- Kristensen LS et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691 (2019). - PubMed
-
- Hulstaert E et al. Charting extracellular transcriptomes in the Human Biofluid RNA Atlas. Cell Rep. 33, 108552 (2020). - PubMed
-
- Vromman M et al. Validation of circular RNAs using RT-qPCR after effective removal of linear RNAs by ribonuclease R. Curr. Protoc. 1, e181 (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
