

SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks
- PMID: 37443338
- PMCID: PMC10482700
- DOI: 10.1038/s41592-023-01938-4
SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks
Abstract
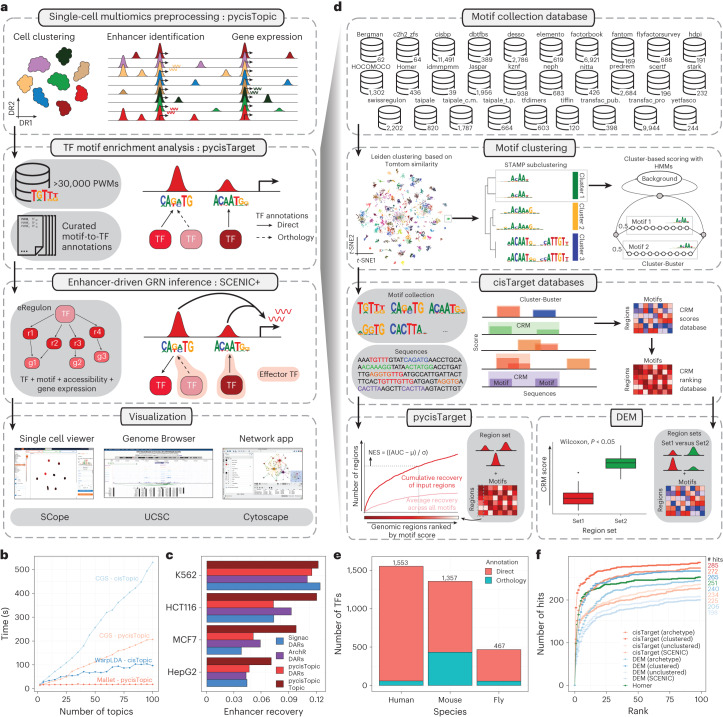
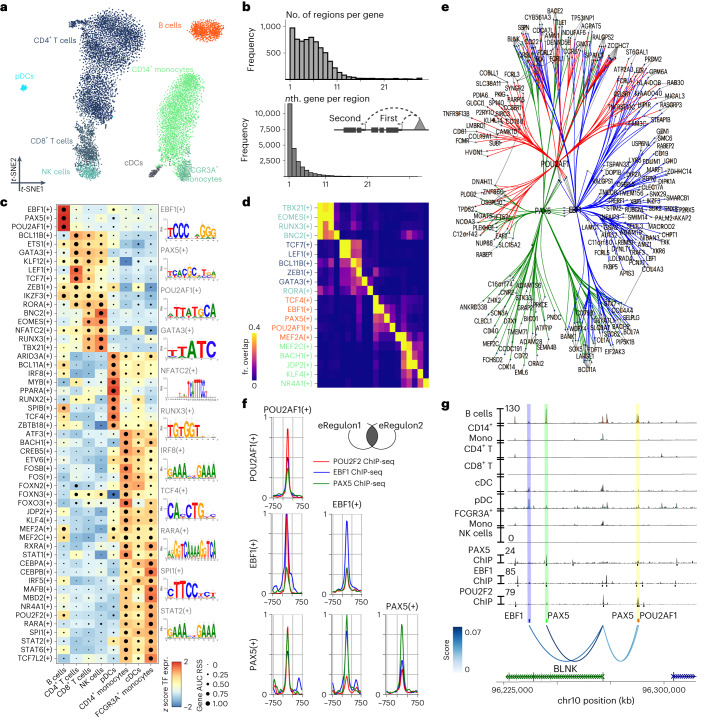
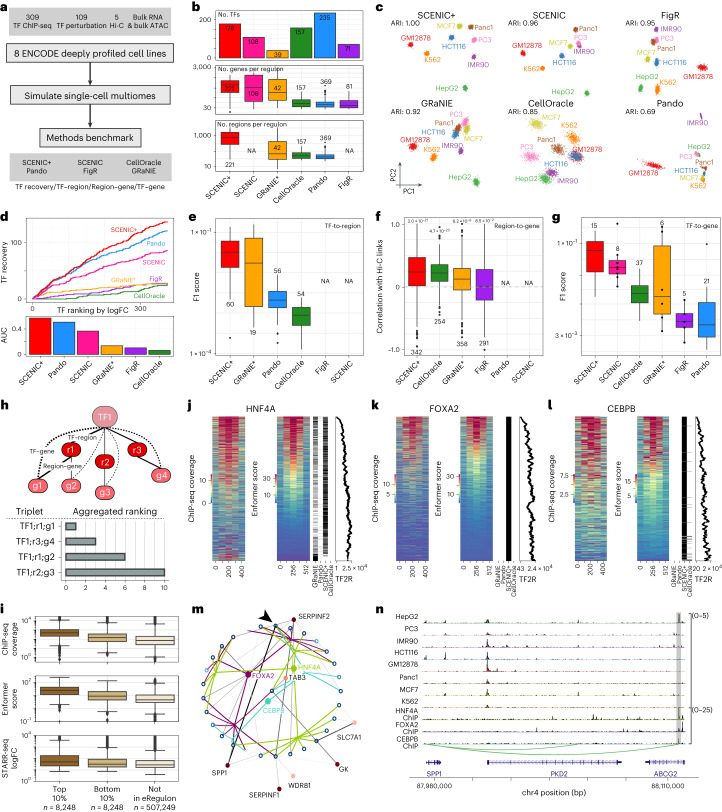
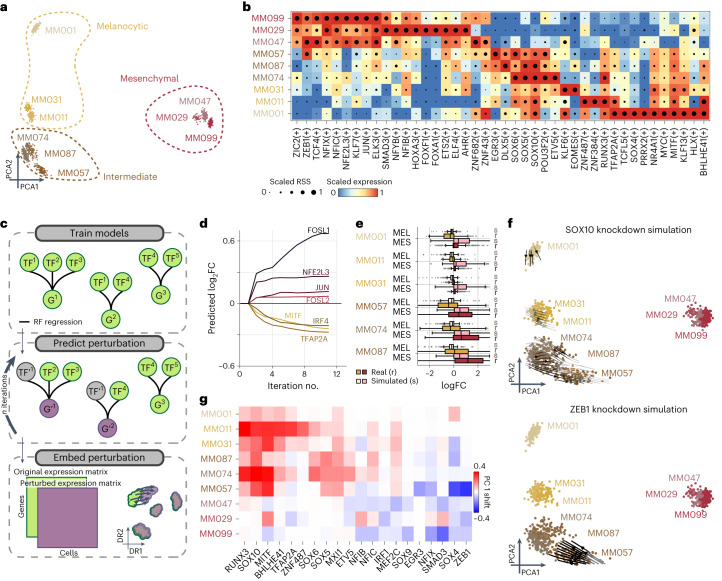
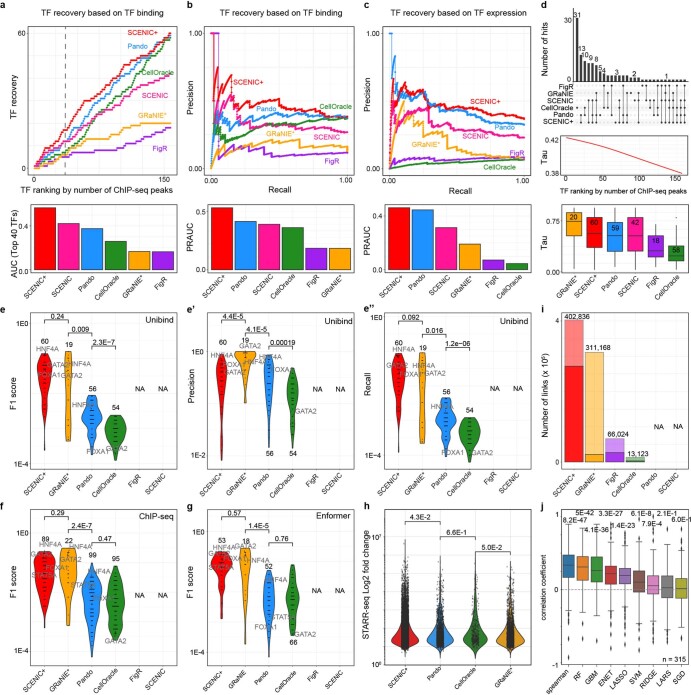
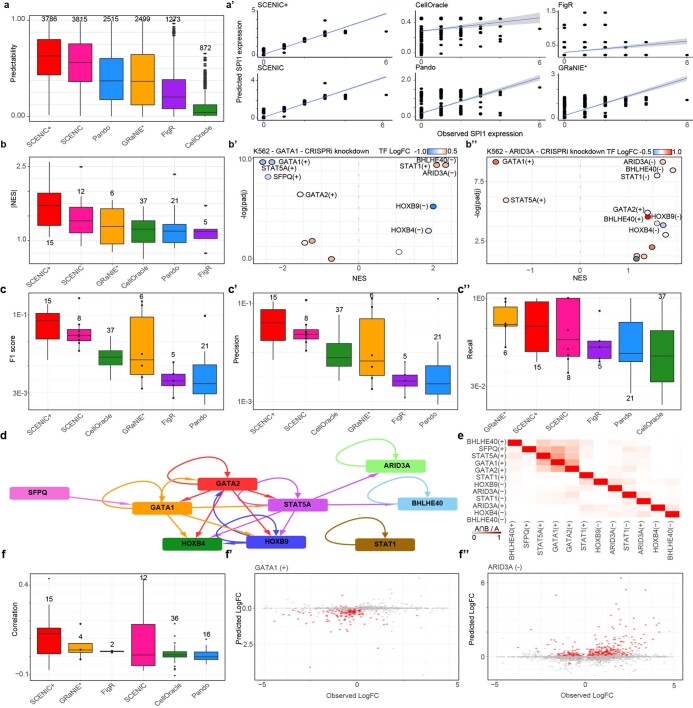
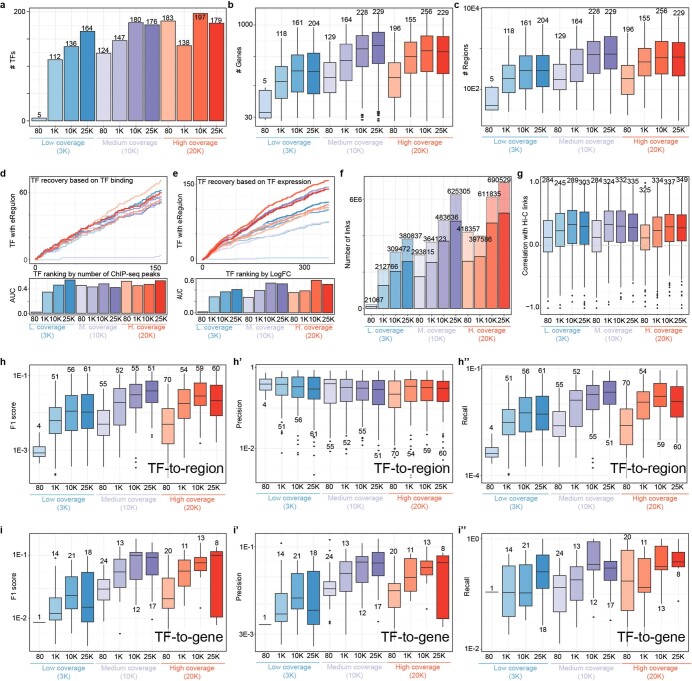
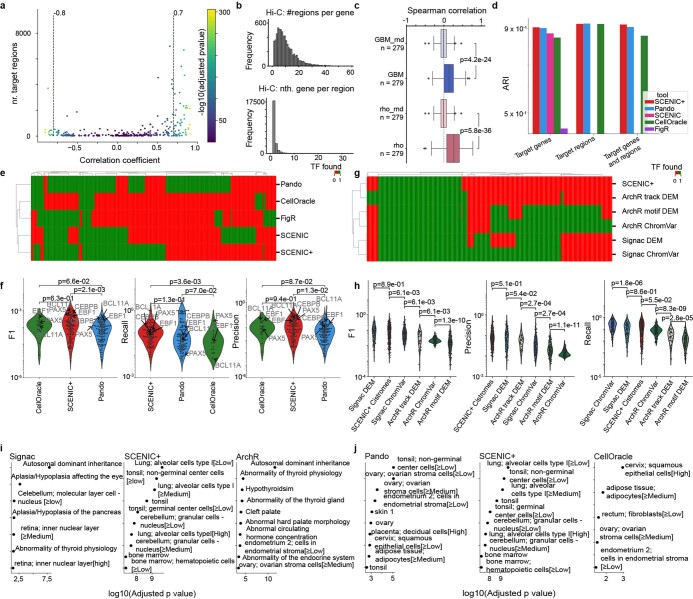
Joint profiling of chromatin accessibility and gene expression in individual cells provides an opportunity to decipher enhancer-driven gene regulatory networks (GRNs). Here we present a method for the inference of enhancer-driven GRNs, called SCENIC+. SCENIC+ predicts genomic enhancers along with candidate upstream transcription factors (TFs) and links these enhancers to candidate target genes. To improve both recall and precision of TF identification, we curated and clustered a motif collection with more than 30,000 motifs. We benchmarked SCENIC+ on diverse datasets from different species, including human peripheral blood mononuclear cells, ENCODE cell lines, melanoma cell states and Drosophila retinal development. Next, we exploit SCENIC+ predictions to study conserved TFs, enhancers and GRNs between human and mouse cell types in the cerebral cortex. Finally, we use SCENIC+ to study the dynamics of gene regulation along differentiation trajectories and the effect of TF perturbations on cell state. SCENIC+ is available at scenicplus.readthedocs.io .
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

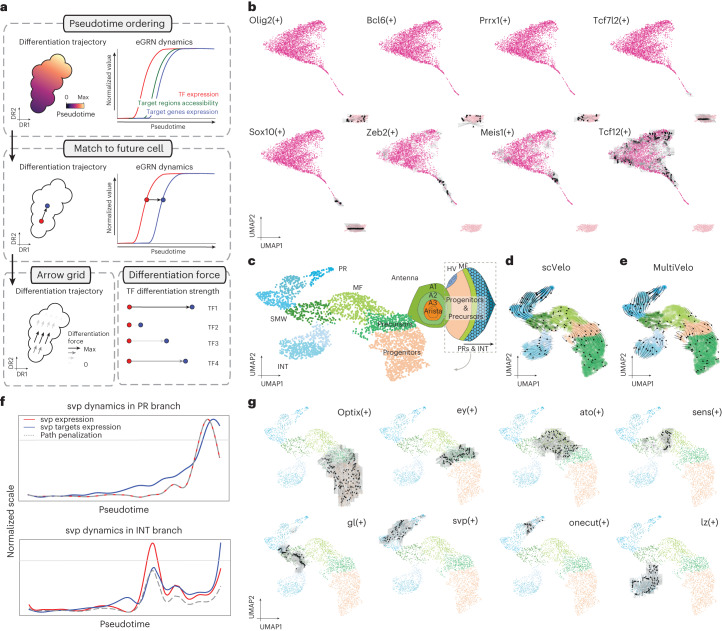
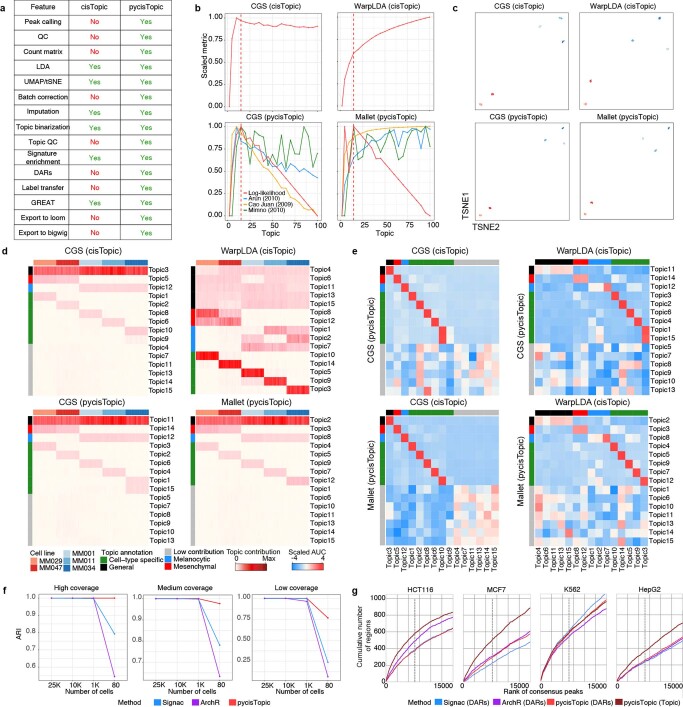
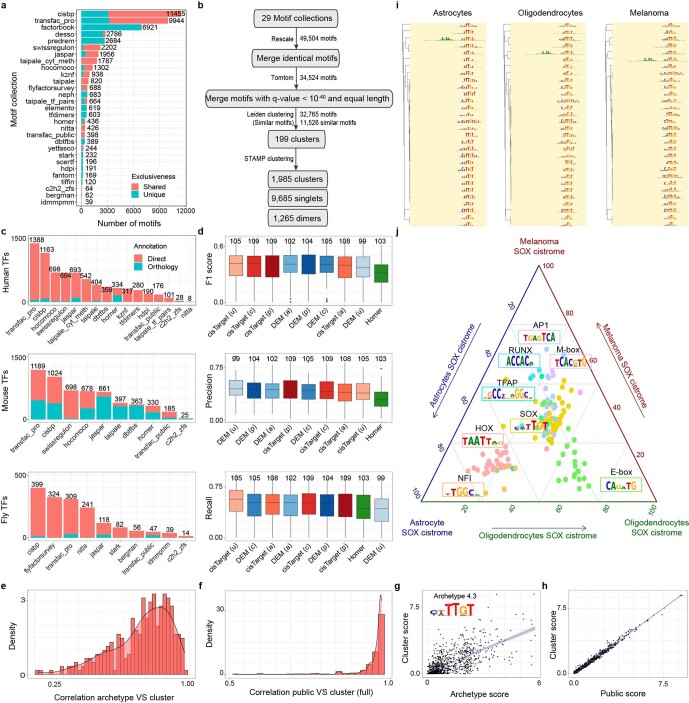
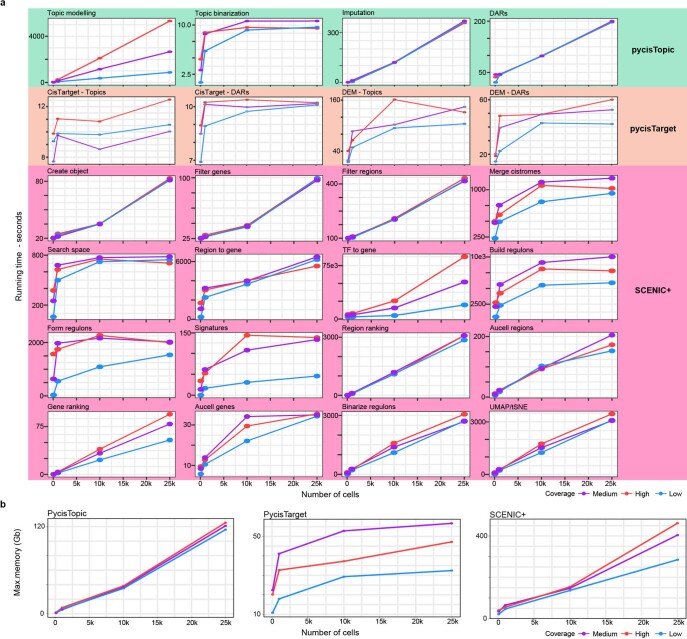
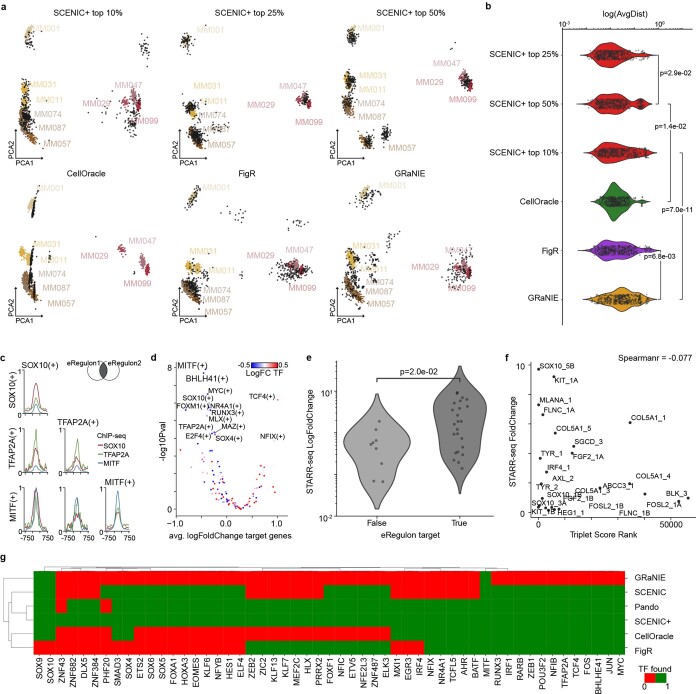
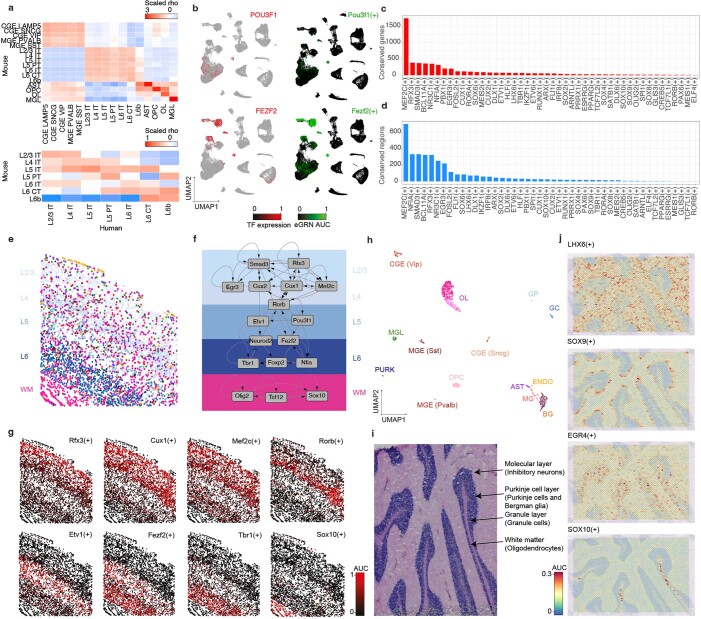
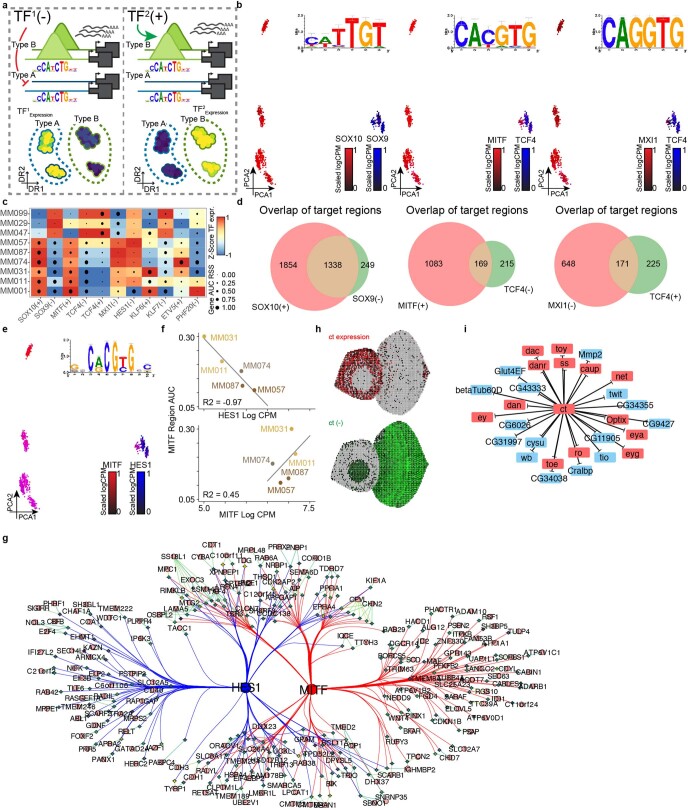
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
