

Investigation of Potential Drug Targets for Cholesterol Regulation to Treat Alzheimer's Disease
- PMID: 37444065
- PMCID: PMC10341567
- DOI: 10.3390/ijerph20136217
Investigation of Potential Drug Targets for Cholesterol Regulation to Treat Alzheimer's Disease
Abstract
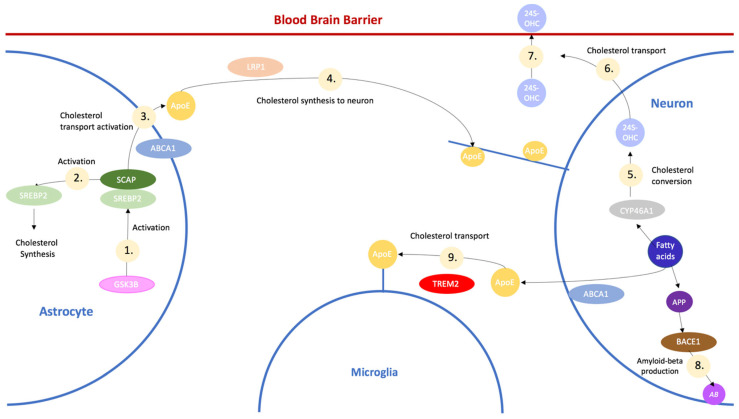
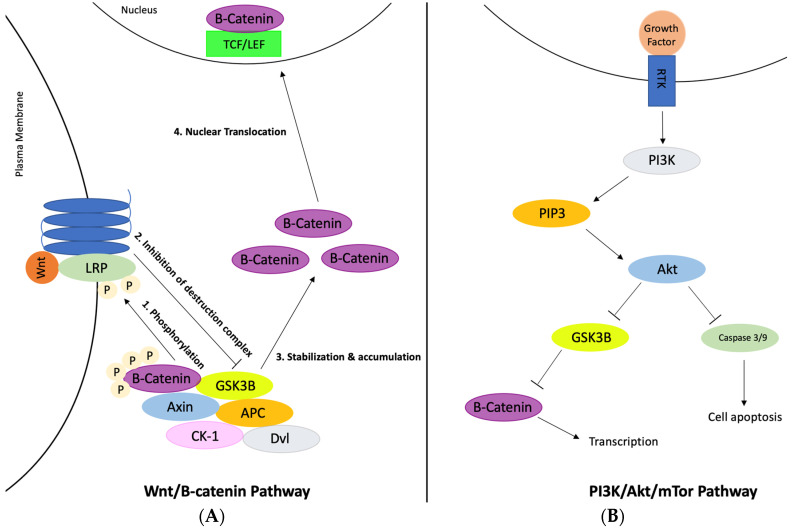
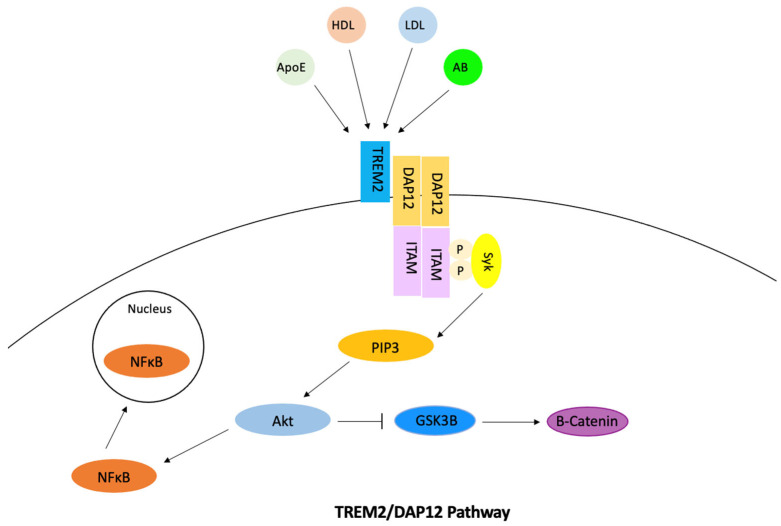
Despite extensive research and seven approved drugs, the complex interplay of genes, proteins, and pathways in Alzheimer's disease remains a challenge. This implies the intricacies of the mechanism for Alzheimer's disease, which involves the interaction of hundreds of genes, proteins, and pathways. While the major hallmarks of Alzheimer's disease are the accumulation of amyloid plaques and tau protein tangles, excessive accumulation of cholesterol is reportedly correlated with Alzheimer's disease patients. In this work, protein-protein interaction analysis was conducted based upon the genes from a clinical database to identify the top protein targets with most data-indicated involvement in Alzheimer's disease, which include ABCA1, CYP46A1, BACE1, TREM2, GSK3B, and SREBP2. The reactions and pathways associated with these genes were thoroughly studied for their roles in regulating brain cholesterol biosynthesis, amyloid beta accumulation, and tau protein tangle formation. Existing clinical trials for each protein target were also investigated. The research indicated that the inhibition of SREBP2, BACE1, or GSK3B is beneficial to reduce cholesterol and amyloid beta accumulation, while the activation of ABCA1, CYP46A1, or TREM2 has similar effects. In this study, Sterol Regulatory Element-Binding Protein 2 (SREBP2) emerged as the primary protein target. SREBP2 serves a pivotal role in maintaining cholesterol balance, acting as a transcription factor that controls the expression of several enzymes pivotal for cholesterol biosynthesis. Novel studies suggest that SREBP2 performs a multifaceted role in Alzheimer's disease. The hyperactivity of SREBP2 may lead to heightened cholesterol biosynthesis, which suggested association with the pathogenesis of Alzheimer's disease. Lowering SREBP2 levels in an Alzheimer's disease mouse model results in reduced production of amyloid-beta, a major contributor to Alzheimer's disease progression. Moreover, its thoroughly analyzed crystal structure allows for computer-aided screening of potential inhibitors; SREBP2 is thus selected as a prospective drug target. While more protein targets can be added onto the list in the future, this work provides an overview of key proteins involved in the regulation of brain cholesterol biosynthesis that may be further investigated for Alzheimer's disease intervention.
Keywords: Alzheimer’s disease; amyloid beta; cholesterol biosynthesis; drug discovery; ligand-protein docking; protein target; protein-protein interaction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Balch B. Recent Breakthroughs in Alzheimer’s Research Provide Hope for Patients. Association of American Medical Colleges. Jan 24, 2023. [(accessed on 1 March 2023)]. Available online: https://www.aamc.org/news/recent-breakthroughs-alzheimer-s-research-prov....
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
