

Role of innate immunity and systemic inflammation in cystic fibrosis disease progression
- PMID: 37449112
- PMCID: PMC10336457
- DOI: 10.1016/j.heliyon.2023.e17553
Role of innate immunity and systemic inflammation in cystic fibrosis disease progression
Abstract
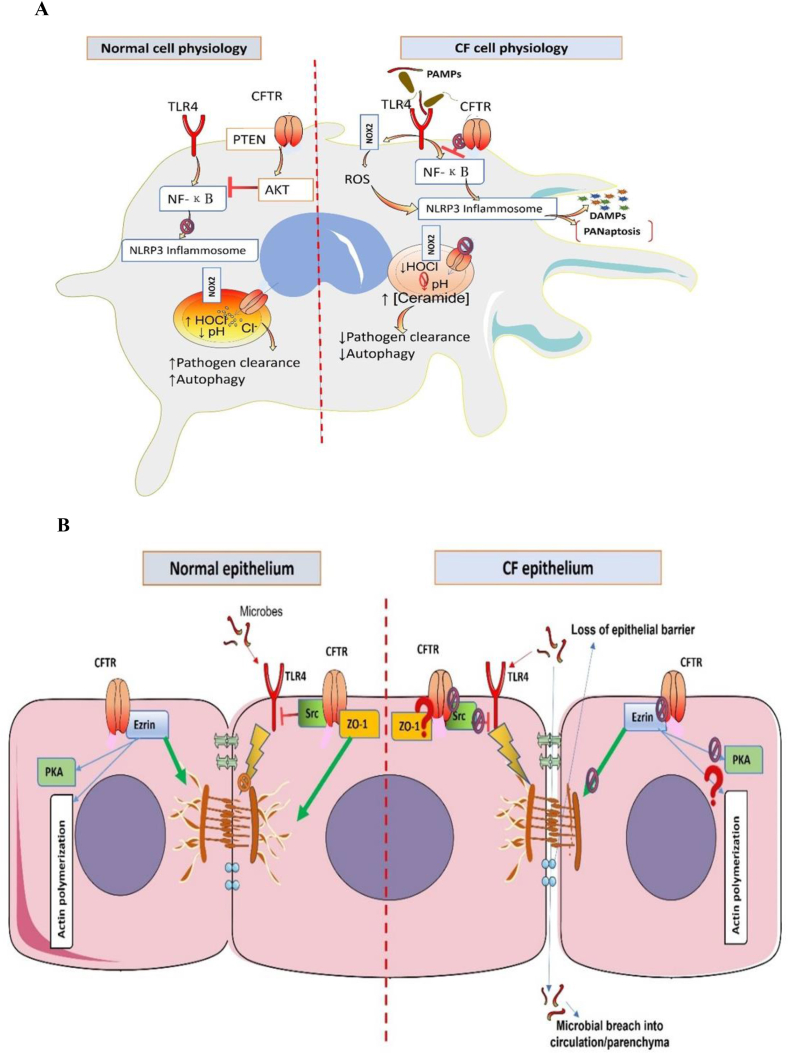
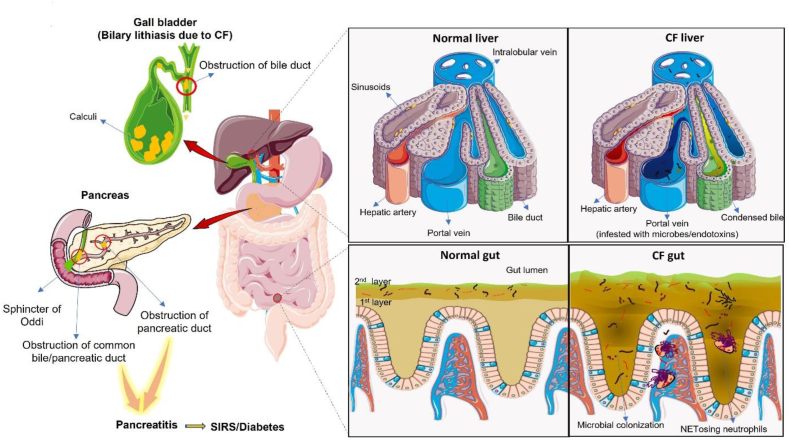
Pathophysiological manifestations of cystic fibrosis (CF) result from a functional defect in the cystic fibrosis transmembrane conductance regulator (CFTR) paving way for mucus obstruction and pathogen colonization. The role of CFTR in modulating immune cell function and vascular integrity, irrespective of mucus thickening, in determining the host cell response to pathogens/allergens and causing systemic inflammation is least appreciated. Since CFTR plays a key role in the conductance of anions like Cl-, loss of CFTR function could affect various basic cellular processes, such as cellular homeostasis, lysosome acidification, and redox balance. CFTR aids in endotoxin tolerance by regulating Toll-like receptor-mediated signaling resulting in uncontrolled activation of innate immune cells. Although leukocytes of CF patients are hyperactivated, they exhibit compromised phagosome activity thus favouring the orchestration of sepsis from defective pathogen clearance. This review will emphasize the importance of innate immunity and systemic inflammatory response in the development of CF and other CFTR-associated pathologies.
Keywords: Cystic fibrosis; Innate immunity; NETosis; PRR-Mediated signaling; Systemic inflammation; T1/T2 immune cell ratio.
© 2023 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures



Similar articles
-
The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity.Hepatology. 2016 Dec;64(6):2118-2134. doi: 10.1002/hep.28817. Epub 2016 Oct 27. Hepatology. 2016. PMID: 27629435 Free PMC article.
-
Innate immune response in CF airway epithelia: hyperinflammatory?Am J Physiol Cell Physiol. 2006 Aug;291(2):C218-30. doi: 10.1152/ajpcell.00605.2005. Am J Physiol Cell Physiol. 2006. PMID: 16825601 Review.
-
Pharmacotherapy of the ion transport defect in cystic fibrosis: role of purinergic receptor agonists and other potential therapeutics.Am J Respir Med. 2003;2(4):299-309. doi: 10.1007/BF03256658. Am J Respir Med. 2003. PMID: 14719996 Review.
-
Revisiting the Role of Leukocytes in Cystic Fibrosis.Cells. 2021 Dec 1;10(12):3380. doi: 10.3390/cells10123380. Cells. 2021. PMID: 34943888 Free PMC article. Review.
-
Immune responses in cystic fibrosis: are they intrinsically defective?Am J Respir Cell Mol Biol. 2012 Jun;46(6):715-22. doi: 10.1165/rcmb.2011-0399RT. Epub 2012 Mar 8. Am J Respir Cell Mol Biol. 2012. PMID: 22403802
Cited by
-
Mechanisms of Lung Cancer Development in Cystic Fibrosis Patients: The Role of Inflammation, Oxidative Stress, and Lung Microbiome Dysbiosis.Biomolecules. 2025 Jun 6;15(6):828. doi: 10.3390/biom15060828. Biomolecules. 2025. PMID: 40563468 Free PMC article. Review.
-
A New Frontier in Cystic Fibrosis Pathophysiology: How and When Clock Genes Can Affect the Inflammatory/Immune Response in a Genetic Disease Model.Curr Issues Mol Biol. 2024 Sep 18;46(9):10396-10410. doi: 10.3390/cimb46090618. Curr Issues Mol Biol. 2024. PMID: 39329970 Free PMC article. Review.
-
Immune Factors, Immune Cells and Inflammatory Diseases.Int J Mol Sci. 2024 Feb 19;25(4):2417. doi: 10.3390/ijms25042417. Int J Mol Sci. 2024. PMID: 38397094 Free PMC article.
-
Nanoparticles as Drug Delivery Vehicles for People with Cystic Fibrosis.Biomimetics (Basel). 2024 Sep 22;9(9):574. doi: 10.3390/biomimetics9090574. Biomimetics (Basel). 2024. PMID: 39329596 Free PMC article. Review.
References
-
- Xu Y., Yuan Q., Cao S., Cui S., Xue L., Song X., Li Z., Xu R., Yuan Q., Li R. Aldehyde dehydrogenase 2 inhibited oxidized LDL-induced NLRP3 inflammasome priming and activation via attenuating oxidative stress. Biochem. Biophys. Res. Commun. 2020;529:998–1004. doi: 10.1016/j.bbrc.2020.06.075. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
