

RAS-dependent RAF-MAPK hyperactivation by pathogenic RIT1 is a therapeutic target in Noonan syndrome-associated cardiac hypertrophy
- PMID: 37450595
- PMCID: PMC10348673
- DOI: 10.1126/sciadv.adf4766
RAS-dependent RAF-MAPK hyperactivation by pathogenic RIT1 is a therapeutic target in Noonan syndrome-associated cardiac hypertrophy
Abstract
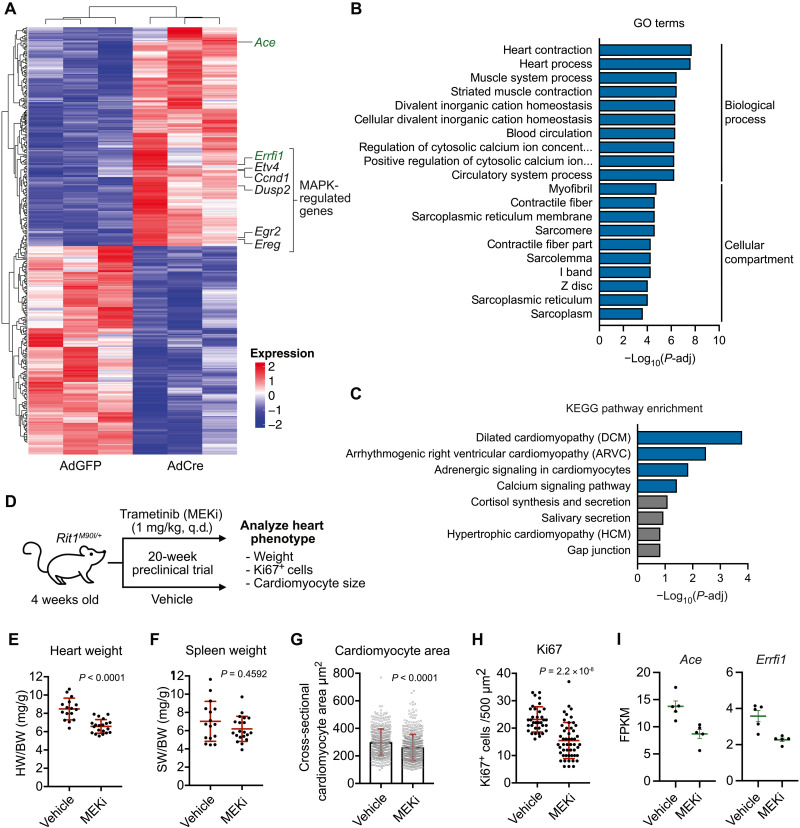
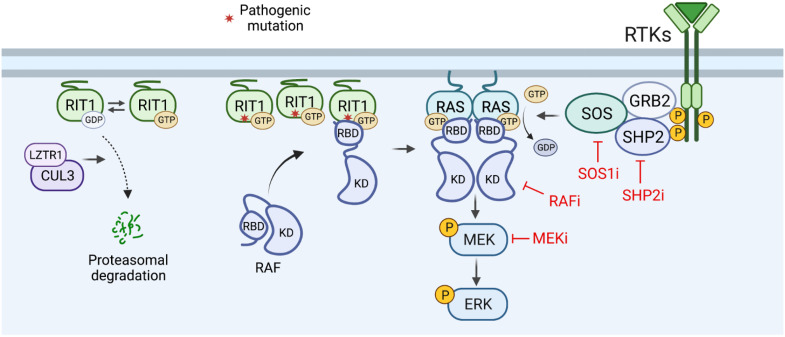
RIT1 is a RAS guanosine triphosphatase (GTPase) that regulates different aspects of signal transduction and is mutated in lung cancer, leukemia, and in the germline of individuals with Noonan syndrome. Pathogenic RIT1 proteins promote mitogen-activated protein kinase (MAPK) hyperactivation; however, this mechanism remains poorly understood. Here, we show that RAF kinases are direct effectors of membrane-bound mutant RIT1 necessary for MAPK activation. We identify critical residues in RIT1 that facilitate interaction with membrane lipids and show that these are necessary for association with RAF kinases and MAPK activation. Although mutant RIT1 binds to RAF kinases directly, it fails to activate MAPK signaling in the absence of classical RAS proteins. Consistent with aberrant RAF/MAPK activation as a driver of disease, we show that pathway inhibition alleviates cardiac hypertrophy in a mouse model of RIT1 mutant Noonan syndrome. These data shed light on the function of pathogenic RIT1 and identify avenues for therapeutic intervention.
Figures
Similar articles
-
Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome.Am J Hum Genet. 2013 Jul 11;93(1):173-80. doi: 10.1016/j.ajhg.2013.05.021. Epub 2013 Jun 20. Am J Hum Genet. 2013. PMID: 23791108 Free PMC article.
-
New Noonan syndrome model mice with RIT1 mutation exhibit cardiac hypertrophy and susceptibility to β-adrenergic stimulation-induced cardiac fibrosis.EBioMedicine. 2019 Apr;42:43-53. doi: 10.1016/j.ebiom.2019.03.014. Epub 2019 Mar 18. EBioMedicine. 2019. PMID: 30898653 Free PMC article.
-
Mutations in RIT1 cause Noonan syndrome - additional functional evidence and expanding the clinical phenotype.Clin Genet. 2016 Mar;89(3):359-66. doi: 10.1111/cge.12608. Epub 2015 Jun 4. Clin Genet. 2016. PMID: 25959749 Free PMC article.
-
Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway.Expert Rev Mol Med. 2008 Dec 9;10:e37. doi: 10.1017/S1462399408000902. Expert Rev Mol Med. 2008. PMID: 19063751 Review.
-
The molecular functions of RIT1 and its contribution to human disease.Biochem J. 2020 Aug 14;477(15):2755-2770. doi: 10.1042/BCJ20200442. Biochem J. 2020. PMID: 32766847 Free PMC article. Review.
Cited by
-
Complex interplay between RAS GTPases and RASSF effectors regulates subcellular localization of YAP.EMBO Rep. 2024 Aug;25(8):3574-3600. doi: 10.1038/s44319-024-00203-9. Epub 2024 Jul 15. EMBO Rep. 2024. PMID: 39009833 Free PMC article.
-
RIT1 Promotes the Proliferation of Gliomas Through the Regulation of the PI3K/AKT/c-Myc Signalling Pathway.J Cell Mol Med. 2025 Jan;29(2):e70362. doi: 10.1111/jcmm.70362. J Cell Mol Med. 2025. PMID: 39833023 Free PMC article.
-
Functional and structural insights into RAS effector proteins.Mol Cell. 2024 Aug 8;84(15):2807-2821. doi: 10.1016/j.molcel.2024.06.027. Epub 2024 Jul 17. Mol Cell. 2024. PMID: 39025071 Free PMC article. Review.
-
The deubiquitinase USP9X regulates RIT1 protein abundance and oncogenic phenotypes.iScience. 2024 Jul 14;27(8):110499. doi: 10.1016/j.isci.2024.110499. eCollection 2024 Aug 16. iScience. 2024. PMID: 39161959 Free PMC article.
-
RIT1 Drives Oncogenic Transformation and is an Actionable Target in Lung Adenocarcinoma.Cancer Res. 2025 Jul 11:10.1158/0008-5472.CAN-24-3819. doi: 10.1158/0008-5472.CAN-24-3819. Online ahead of print. Cancer Res. 2025. PMID: 40644578 Free PMC article.
References
-
- S. J. Leevers, H. F. Paterson, C. J. Marshall, Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369, 411–414 (1994). - PubMed
-
- D. Stokoe, S. G. Macdonald, K. Cadwallader, M. Symons, J. F. Hancock, Activation of Raf as a result of recruitment to the plasma membrane. Science 264, 1463–1467 (1994). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
