

Dose imbalance of DYRK1A kinase causes systemic progeroid status in Down syndrome by increasing the un-repaired DNA damage and reducing LaminB1 levels
- PMID: 37451904
- PMCID: PMC10435767
- DOI: 10.1016/j.ebiom.2023.104692
Dose imbalance of DYRK1A kinase causes systemic progeroid status in Down syndrome by increasing the un-repaired DNA damage and reducing LaminB1 levels
Abstract
Background: People with Down syndrome (DS) show clinical signs of accelerated ageing. Causative mechanisms remain unknown and hypotheses range from the (essentially untreatable) amplified-chromosomal-instability explanation, to potential actions of individual supernumerary chromosome-21 genes. The latter explanation could open a route to therapeutic amelioration if the specific over-acting genes could be identified and their action toned-down.
Methods: Biological age was estimated through patterns of sugar molecules attached to plasma immunoglobulin-G (IgG-glycans, an established "biological-ageing-clock") in n = 246 individuals with DS from three European populations, clinically characterised for the presence of co-morbidities, and compared to n = 256 age-, sex- and demography-matched healthy controls. Isogenic human induced pluripotent stem cell (hiPSCs) models of full and partial trisomy-21 with CRISPR-Cas9 gene editing and two kinase inhibitors were studied prior and after differentiation to cerebral organoids.
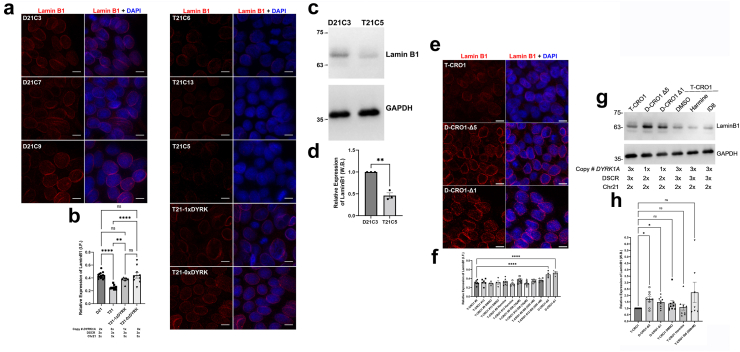
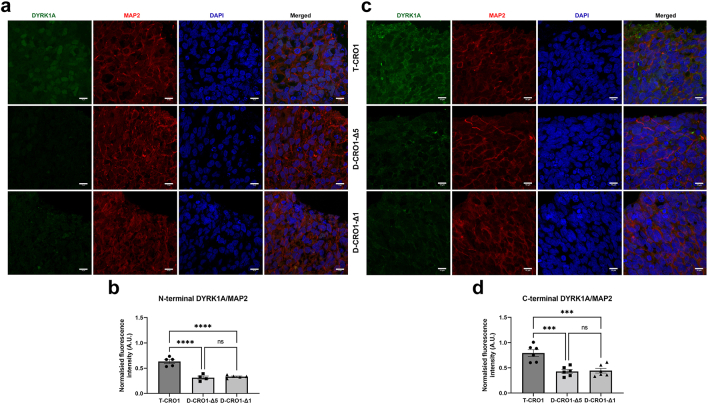
Findings: Biological age in adults with DS is (on average) 18.4-19.1 years older than in chronological-age-matched controls independent of co-morbidities, and this shift remains constant throughout lifespan. Changes are detectable from early childhood, and do not require a supernumerary chromosome, but are seen in segmental duplication of only 31 genes, along with increased DNA damage and decreased levels of LaminB1 in nucleated blood cells. We demonstrate that these cell-autonomous phenotypes can be gene-dose-modelled and pharmacologically corrected in hiPSCs and derived cerebral organoids. Using isogenic hiPSC models we show that chromosome-21 gene DYRK1A overdose is sufficient and necessary to cause excess unrepaired DNA damage.
Interpretation: Explanation of hitherto observed accelerated ageing in DS as a developmental progeroid syndrome driven by DYRK1A overdose provides a target for early pharmacological preventative intervention strategies.
Funding: Main funding came from the "Research Cooperability" Program of the Croatian Science Foundation funded by the European Union from the European Social Fund under the Operational Programme Efficient Human Resources 2014-2020, Project PZS-2019-02-4277, and the Wellcome Trust Grants 098330/Z/12/Z and 217199/Z/19/Z (UK). All other funding is described in details in the "Acknowledgements".
Keywords: Ageing; Chromosome 21; DYRK1A; DYRK1A inhibitors; Down syndrome; Down syndrome critical region; IgG glycan; LaminB1.
Copyright © 2023 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests GL is the founder and owner of Genos Ltd., a private research organisation that specialises in high-throughput glycomic analyses and has several patents in this field and is also a shareholder in GlycanAge Ltd., a company that sells the GlycanAge test of biological age. AC, FV, JJ, MPB, ASla, HD, AF, DP and JK are employees of Genos Ltd. AStr has served on the Advisory Boards of AC Immune and ProMIS Neuroscience, and is a past president of the Trisomy21 Research Society. TS is the scientific co-founder and a shareholder of Zoe Ltd.
Figures


















References
-
- Cuadrado E., Barrena M.J. Immune dysfunction in Down's syndrome: primary immune deficiency or early senescence of the immune system? Clin Immunol Immunopathol. 1996;78(3):209–214. - PubMed
-
- da Silva V.Z., de Franca Barros J., de Azevedo M., de Godoy J.R., Arena R., Cipriano G., Jr. Bone mineral density and respiratory muscle strength in male individuals with mental retardation (with and without Down Syndrome) Res Dev Disabil. 2010;31(6):1585–1589. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
