

The complete and fully-phased diploid genome of a male Han Chinese
- PMID: 37452091
- PMCID: PMC10542383
- DOI: 10.1038/s41422-023-00849-5
The complete and fully-phased diploid genome of a male Han Chinese
Abstract
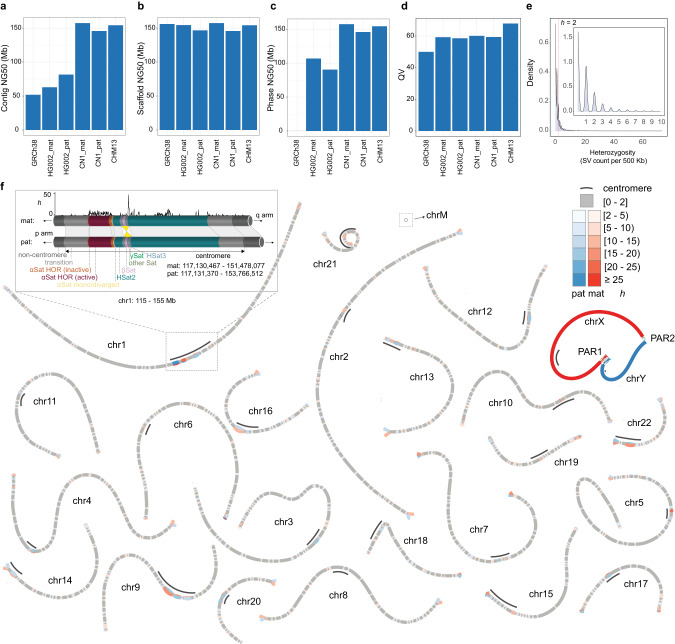
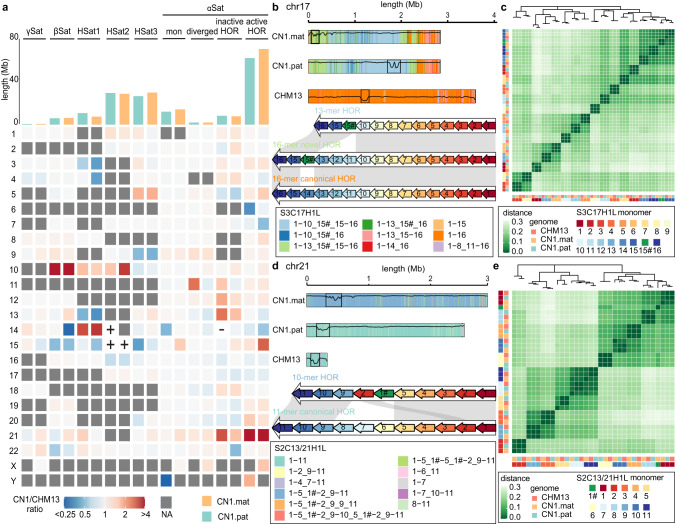
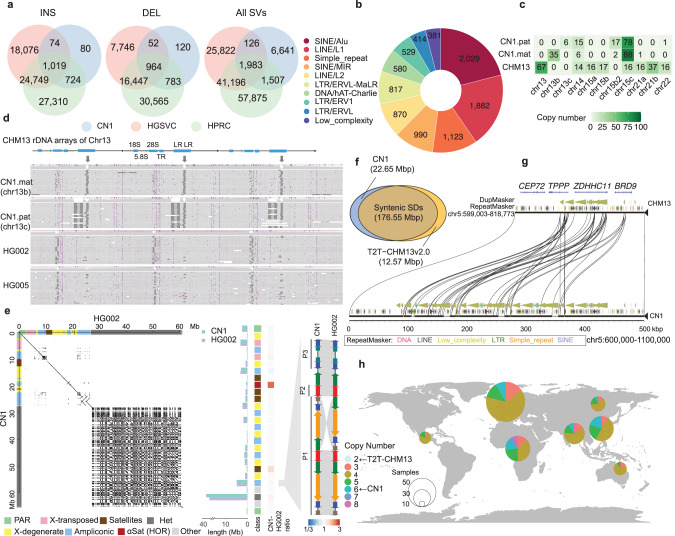
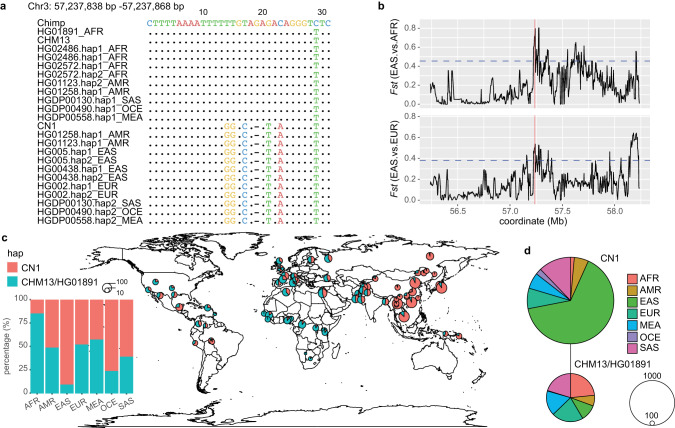
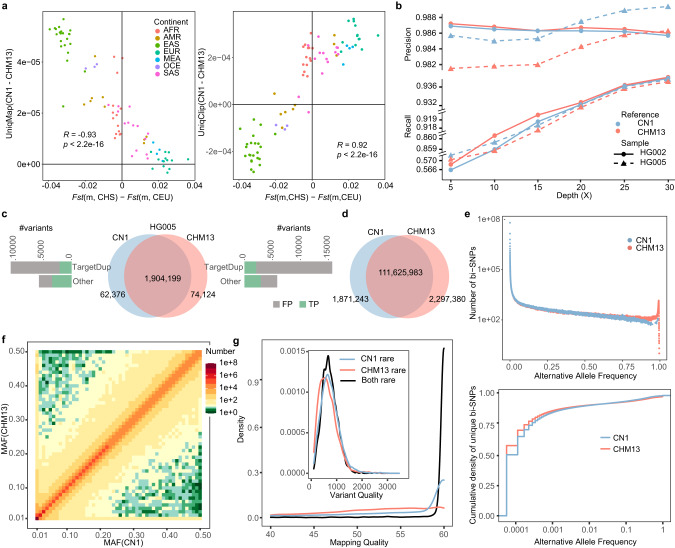
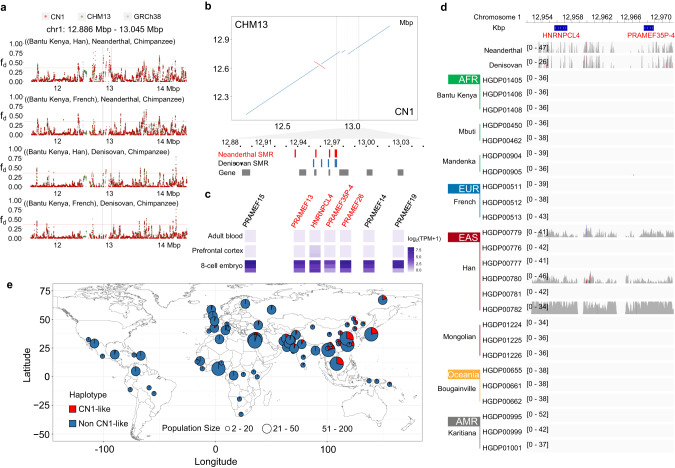
Since the release of the complete human genome, the priority of human genomic study has now been shifting towards closing gaps in ethnic diversity. Here, we present a fully phased and well-annotated diploid human genome from a Han Chinese male individual (CN1), in which the assemblies of both haploids achieve the telomere-to-telomere (T2T) level. Comparison of this diploid genome with the CHM13 haploid T2T genome revealed significant variations in the centromere. Outside the centromere, we discovered 11,413 structural variations, including numerous novel ones. We also detected thousands of CN1 alleles that have accumulated high substitution rates and a few that have been under positive selection in the East Asian population. Further, we found that CN1 outperforms CHM13 as a reference genome in mapping and variant calling for the East Asian population owing to the distinct structural variants of the two references. Comparison of SNP calling for a large cohort of 8869 Chinese genomes using CN1 and CHM13 as reference respectively showed that the reference bias profoundly impacts rare SNP calling, with nearly 2 million rare SNPs miss-called with different reference genomes. Finally, applying the CN1 as a reference, we discovered 5.80 Mb and 4.21 Mb putative introgression sequences from Neanderthal and Denisovan, respectively, including many East Asian specific ones undetected using CHM13 as the reference. Our analyses reveal the advances of using CN1 as a reference for population genomic studies and paleo-genomic studies. This complete genome will serve as an alternative reference for future genomic studies on the East Asian population.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
