

ATR protects ongoing and newly assembled DNA replication forks through distinct mechanisms
- PMID: 37454295
- PMCID: PMC10529362
- DOI: 10.1016/j.celrep.2023.112792
ATR protects ongoing and newly assembled DNA replication forks through distinct mechanisms
Abstract
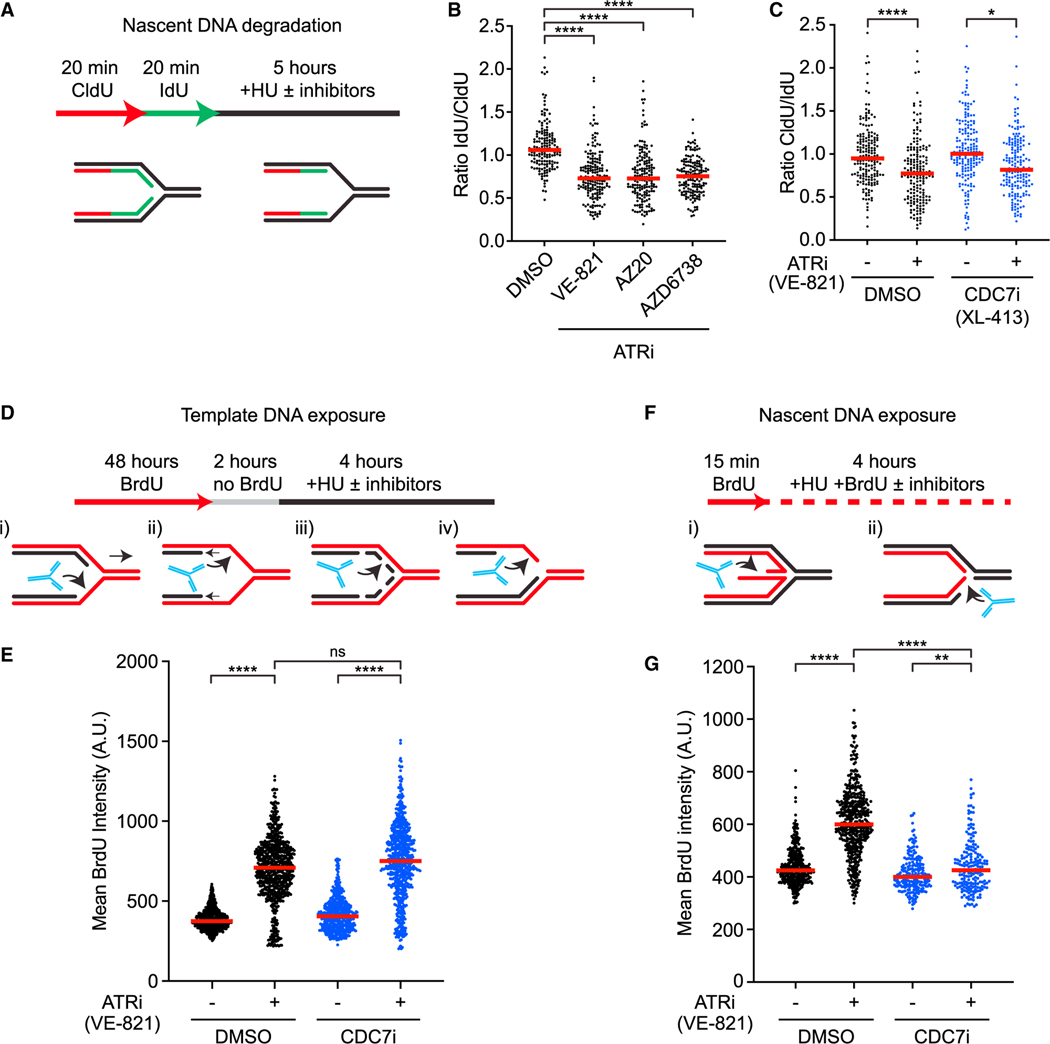
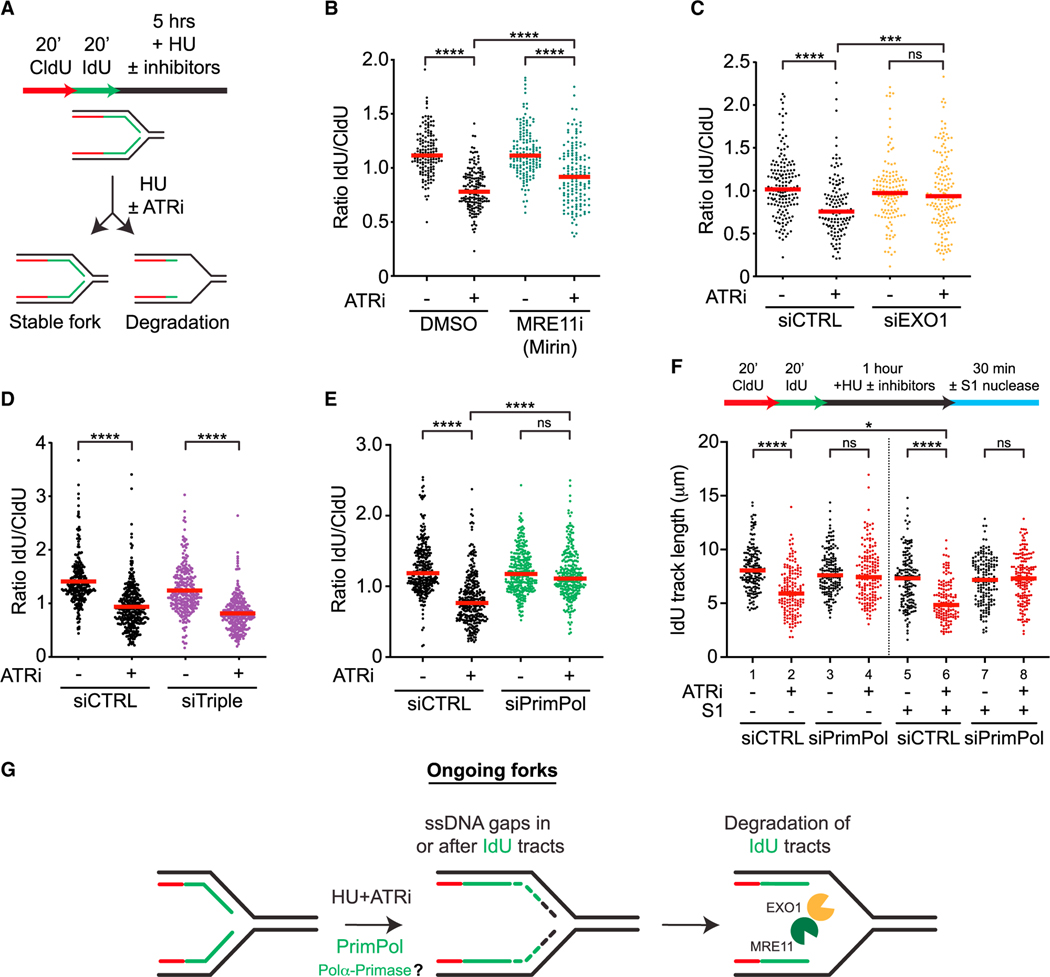
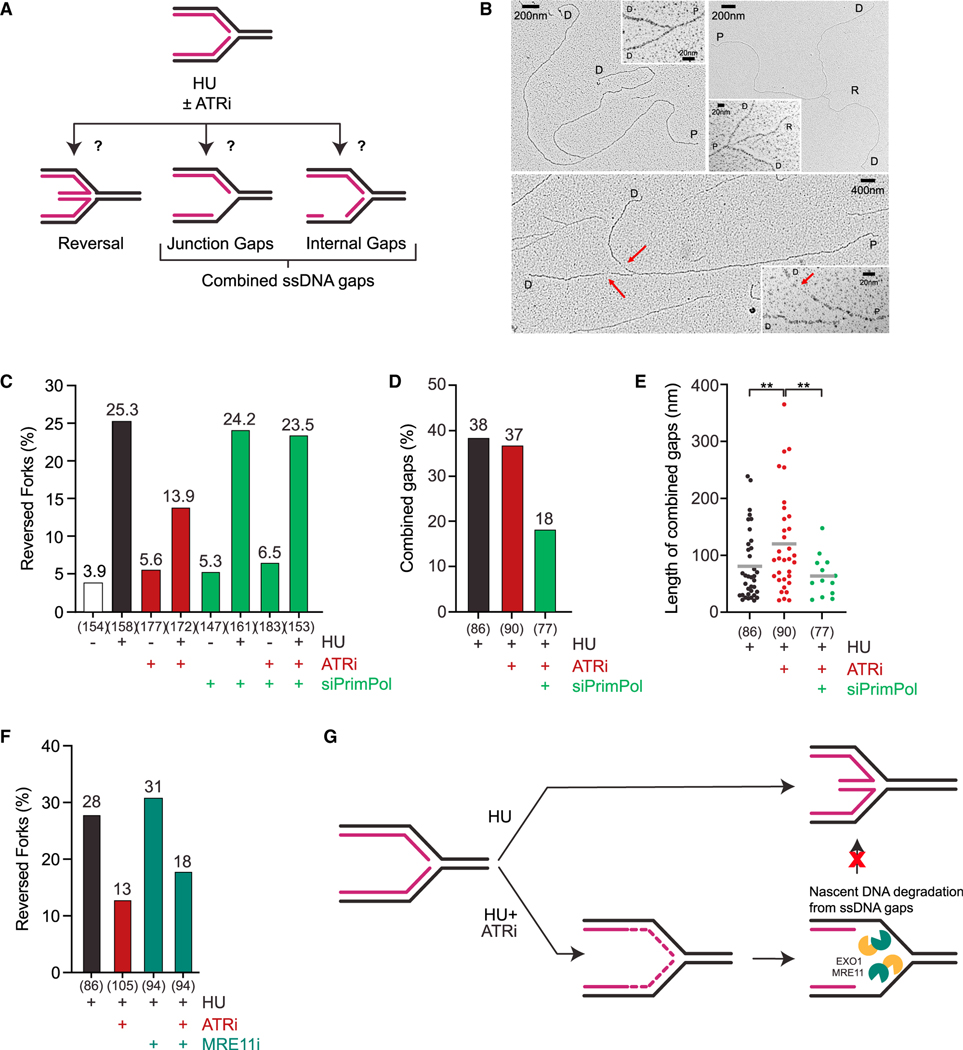
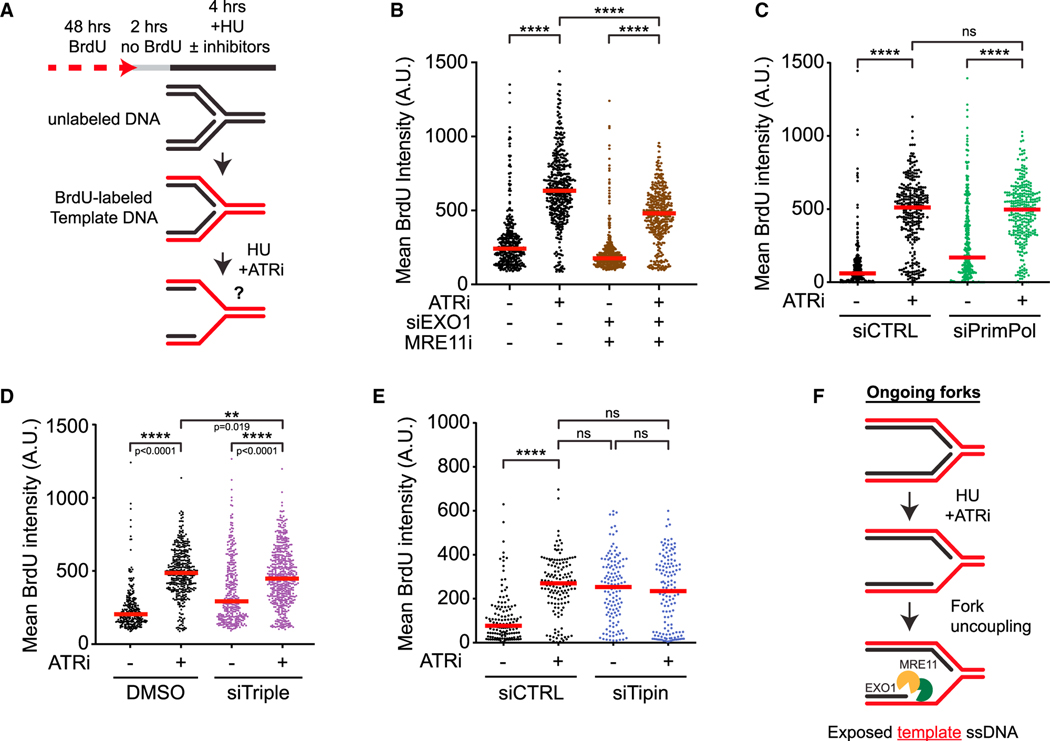
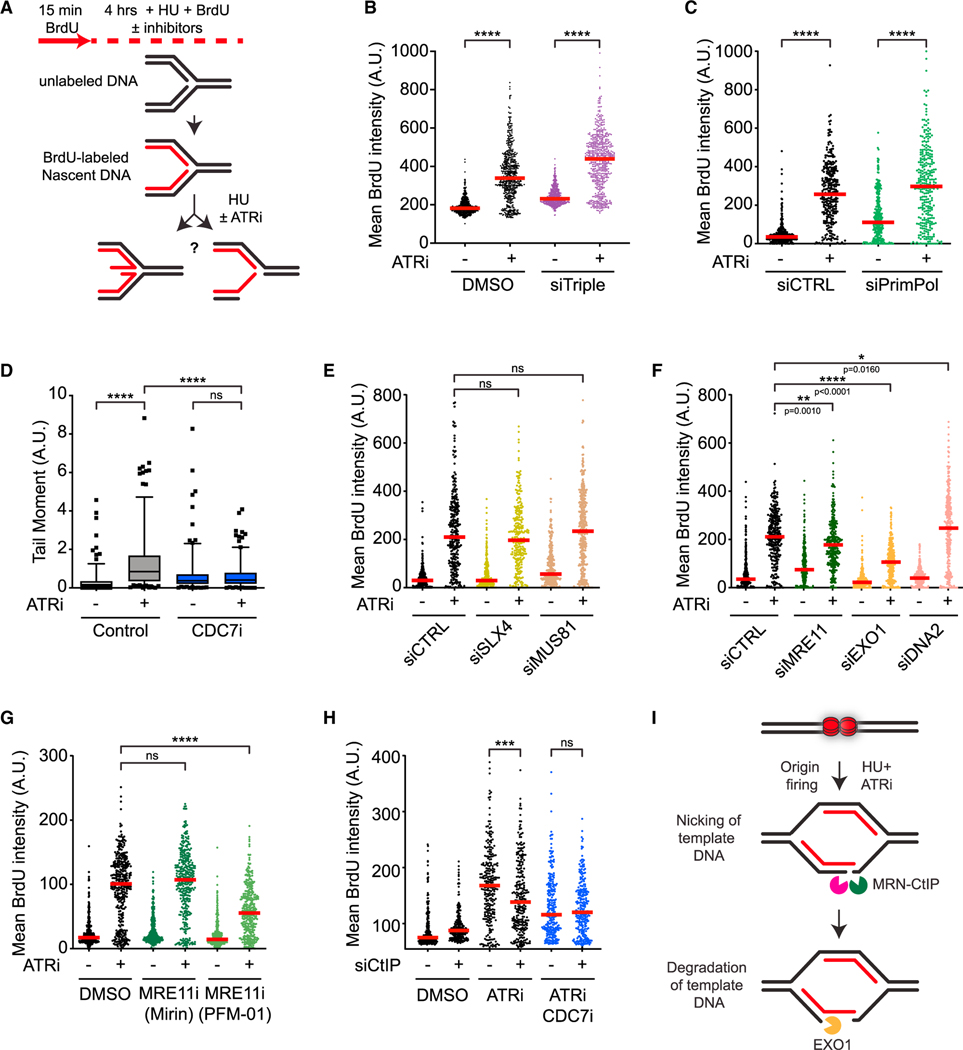
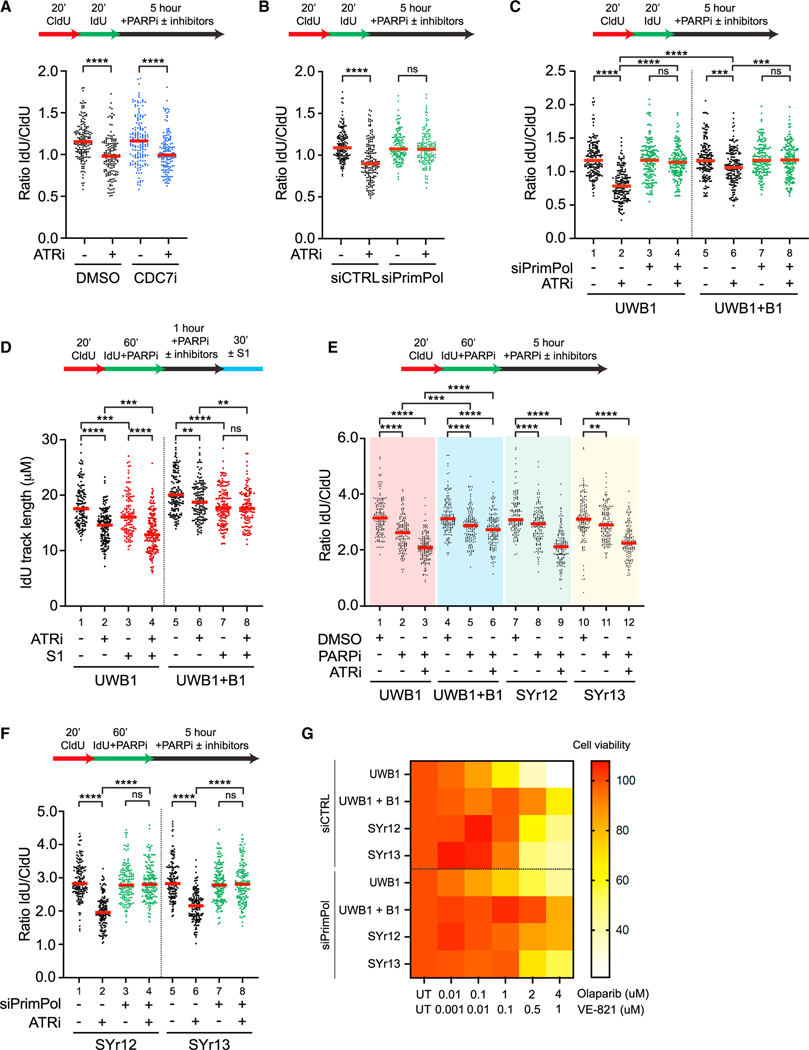
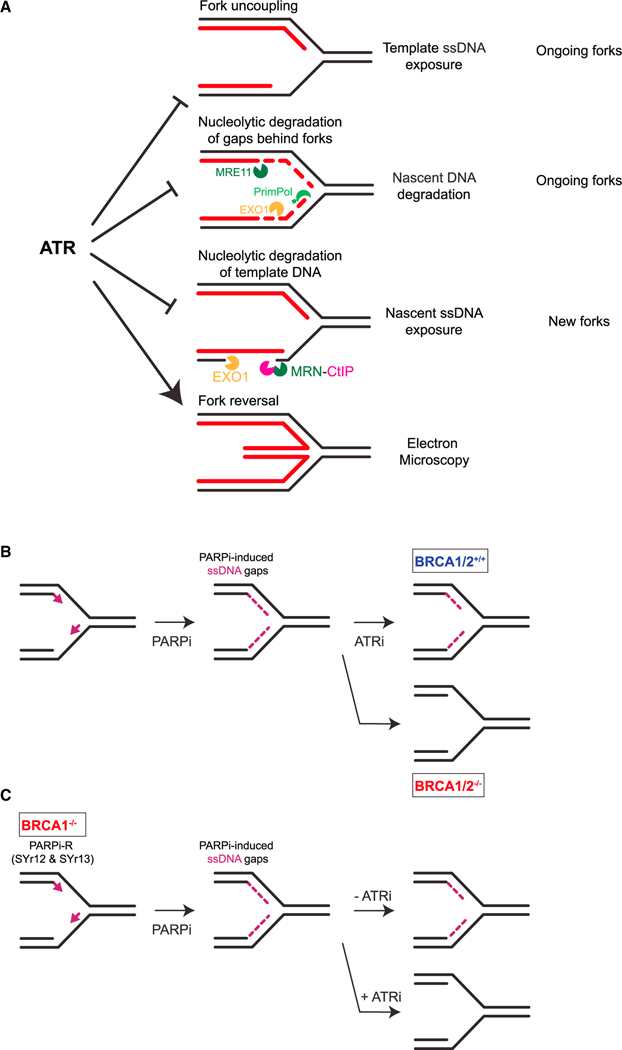
The ATR kinase safeguards genomic integrity during S phase, but how ATR protects DNA replication forks remains incompletely understood. Here, we combine four distinct assays to analyze ATR functions at ongoing and newly assembled replication forks upon replication inhibition by hydroxyurea. At ongoing forks, ATR inhibitor (ATRi) increases MRE11- and EXO1-mediated nascent DNA degradation from PrimPol-generated, single-stranded DNA (ssDNA) gaps. ATRi also exposes template ssDNA through fork uncoupling and nascent DNA degradation. Electron microscopy reveals that ATRi reduces reversed forks by increasing gap-dependent nascent DNA degradation. At new forks, ATRi triggers MRE11- and CtIP-initiated template DNA degradation by EXO1, exposing nascent ssDNA. Upon PARP inhibition, ATRi preferentially exacerbates gap-dependent nascent DNA degradation at ongoing forks in BRCA1/2-deficient cells and disrupts the restored gap protection in BRCA1-deficient, PARP-inhibitor-resistant cells. Thus, ATR protects ongoing and new forks through distinct mechanisms, providing an extended view of ATR's functions in stabilizing replication forks.
Keywords: ATR; BRCA; CP: Molecular biology; PARP inhibitor; gaps; replication; replication fork; single-stranded DNA.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Similar articles
-
PCAF-Mediated Histone Acetylation Promotes Replication Fork Degradation by MRE11 and EXO1 in BRCA-Deficient Cells.Mol Cell. 2020 Oct 15;80(2):327-344.e8. doi: 10.1016/j.molcel.2020.08.018. Epub 2020 Sep 22. Mol Cell. 2020. PMID: 32966758 Free PMC article.
-
MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells.Nat Commun. 2017 Oct 16;8(1):860. doi: 10.1038/s41467-017-01180-5. Nat Commun. 2017. PMID: 29038425 Free PMC article.
-
EXO1 and DNA2-mediated ssDNA gap expansion is essential for ATR activation and to maintain viability in BRCA1-deficient cells.Nucleic Acids Res. 2024 Jun 24;52(11):6376-6391. doi: 10.1093/nar/gkae317. Nucleic Acids Res. 2024. PMID: 38721777 Free PMC article.
-
"Bridging the DNA divide": Understanding the interplay between replication- gaps and homologous recombination proteins RAD51 and BRCA1/2.DNA Repair (Amst). 2024 Sep;141:103738. doi: 10.1016/j.dnarep.2024.103738. Epub 2024 Jul 29. DNA Repair (Amst). 2024. PMID: 39084178 Review.
-
Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51.FEBS Lett. 2017 Apr;591(8):1083-1100. doi: 10.1002/1873-3468.12556. Epub 2017 Jan 30. FEBS Lett. 2017. PMID: 28079255 Review.
Cited by
-
Mechanisms and regulation of replication fork reversal.DNA Repair (Amst). 2024 Sep;141:103731. doi: 10.1016/j.dnarep.2024.103731. Epub 2024 Jul 22. DNA Repair (Amst). 2024. PMID: 39089193 Review.
-
PARP inhibition-associated heterochromatin confers increased DNA replication stress and vulnerability to ATR inhibition in SMARCA4-deficient cells.Cell Death Discov. 2025 Jan 28;11(1):31. doi: 10.1038/s41420-025-02306-1. Cell Death Discov. 2025. PMID: 39875375 Free PMC article.
-
Targeting ATR in patients with cancer.Nat Rev Clin Oncol. 2024 Apr;21(4):278-293. doi: 10.1038/s41571-024-00863-5. Epub 2024 Feb 20. Nat Rev Clin Oncol. 2024. PMID: 38378898 Review.
-
S-phase checkpoint protects from aberrant replication fork processing and degradation.Nucleic Acids Res. 2025 Jul 19;53(14):gkaf707. doi: 10.1093/nar/gkaf707. Nucleic Acids Res. 2025. PMID: 40737093 Free PMC article.
-
The nucleolar protein GNL3 prevents resection of stalled replication forks.EMBO Rep. 2023 Dec 6;24(12):e57585. doi: 10.15252/embr.202357585. Epub 2023 Nov 15. EMBO Rep. 2023. PMID: 37965896 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
