

TBCK syndrome: a rare multi-organ neurodegenerative disease
- PMID: 37455236
- PMCID: PMC10868401
- DOI: 10.1016/j.molmed.2023.06.009
TBCK syndrome: a rare multi-organ neurodegenerative disease
Abstract
TBCK syndrome is an autosomal recessive disorder primarily characterized by global developmental delay, hypotonia, abnormal magnetic resonance imaging (MRI), and distinctive craniofacial phenotypes. High variability is observed among affected individuals and their corresponding variants, making clinical diagnosis challenging. Here, we discuss recent breakthroughs in clinical considerations, TBCK function, and therapeutic development.
Keywords: bone abnormalities; developmental delay; hypotonia; neurodegeneration; neurological disorder; respiratory complications.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures
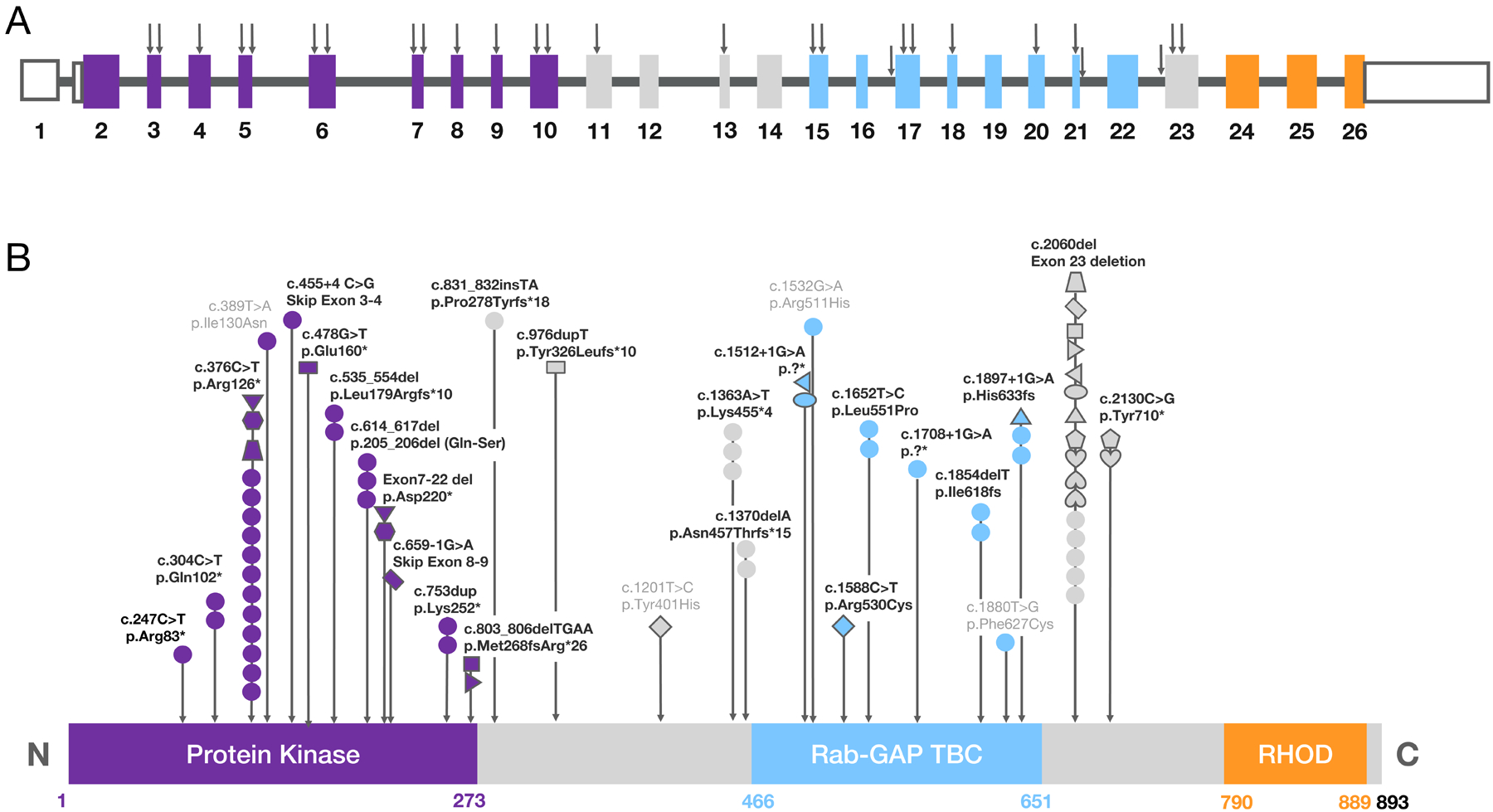
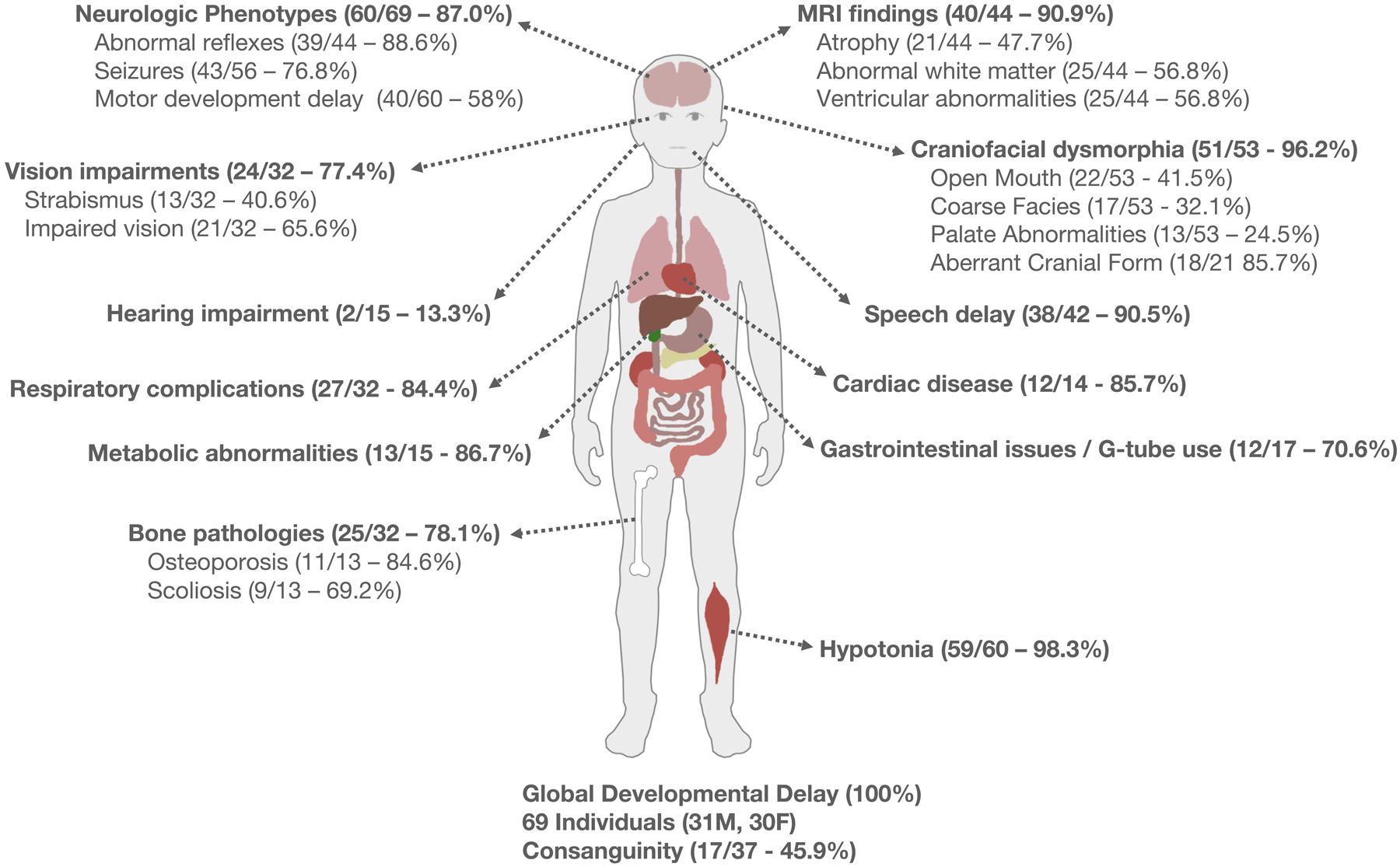
References
-
- Sabanathan S et al. (2023) Expanding the phenotype of children presenting with hypoventilation with biallelic TBCK pathogenic variants and literature review. Neuromuscul. Disord 33, 50–57 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
