

VNtyper enables accurate alignment-free genotyping of MUC1 coding VNTR using short-read sequencing data in autosomal dominant tubulointerstitial kidney disease
- PMID: 37456840
- PMCID: PMC10338300
- DOI: 10.1016/j.isci.2023.107171
VNtyper enables accurate alignment-free genotyping of MUC1 coding VNTR using short-read sequencing data in autosomal dominant tubulointerstitial kidney disease
Abstract
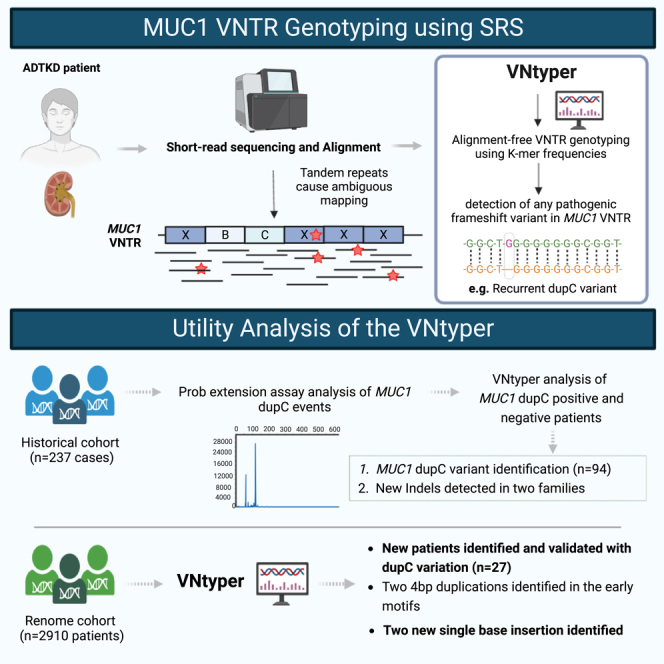
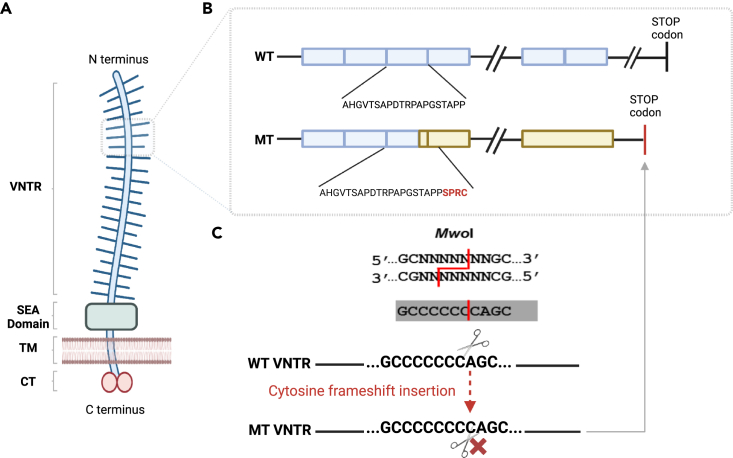
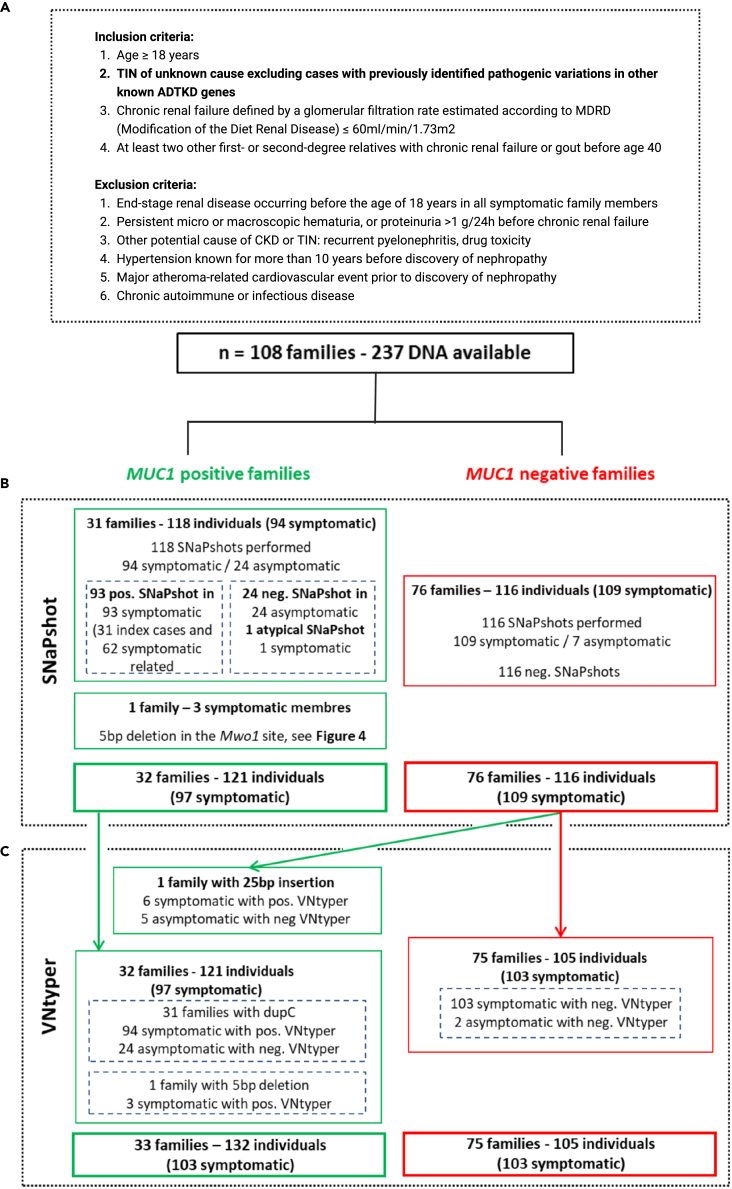
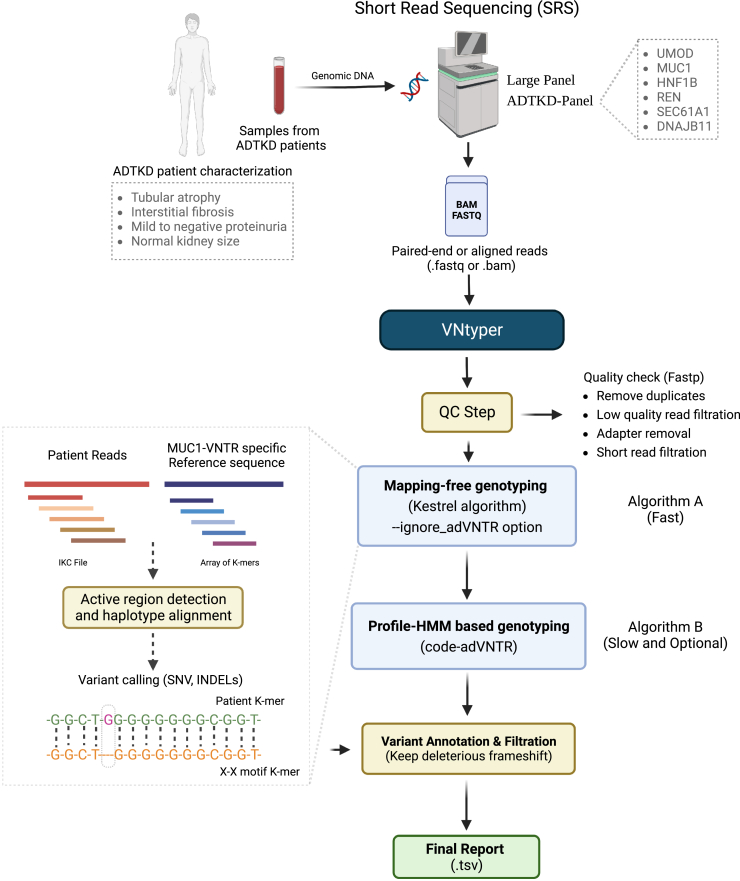
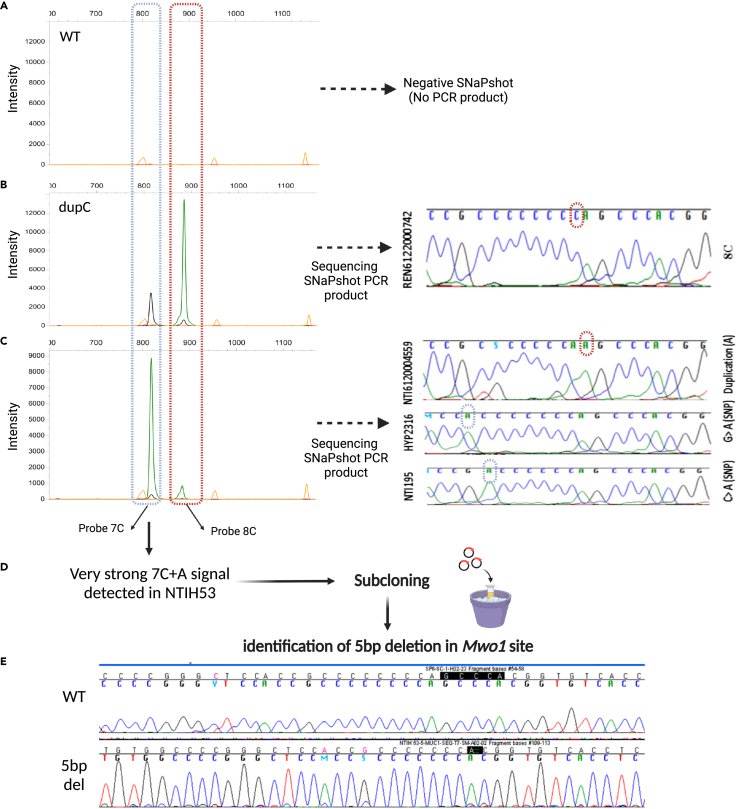
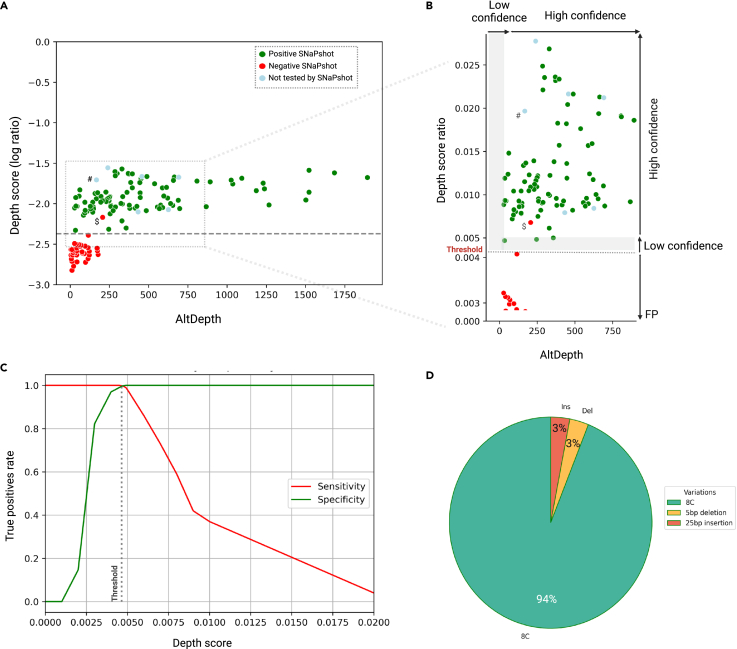
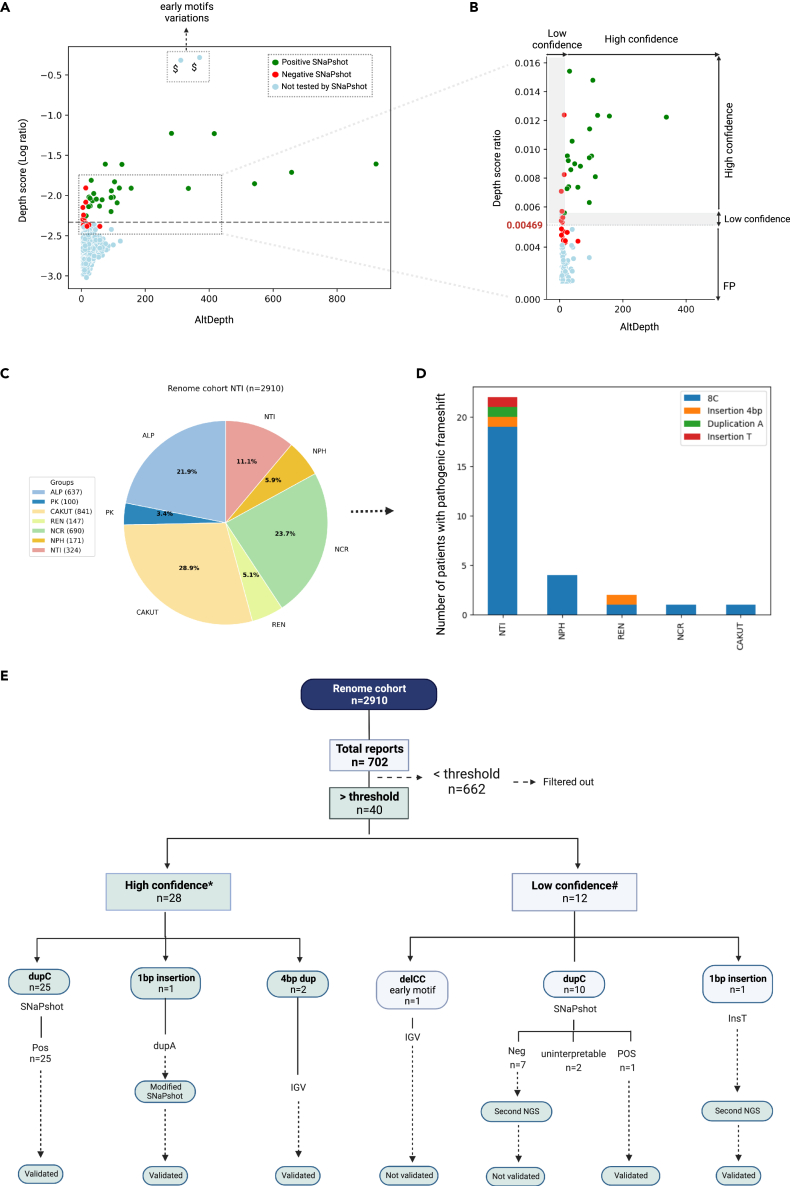
The human genome comprises approximately 3% of tandem repeats with variable length (VNTR), a few of which have been linked to human rare diseases. Autosomal dominant tubulointerstitial kidney disease-MUC1 (ADTKD-MUC1) is caused by specific frameshift variants in the coding VNTR of the MUC1 gene. Calling variants from VNTR using short-read sequencing (SRS) is challenging due to poor read mappability. We developed a computational pipeline, VNtyper, for reliable detection of MUC1 VNTR pathogenic variants and demonstrated its clinical utility in two distinct cohorts: (1) a historical cohort including 108 families with ADTKD and (2) a replication naive cohort comprising 2,910 patients previously tested on a panel of genes involved in monogenic renal diseases. In the historical cohort all cases known to carry pathogenic MUC1 variants were re-identified, and a new 25bp-frameshift insertion in an additional mislaid family was detected. In the replication cohort, we discovered and validated 30 new patients.
Keywords: Genetics; Genomics; Genotyping; Techniques in genetics.
© 2023 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Eckardt K.-U., Alper S.L., Antignac C., Bleyer A.J., Chauveau D., Dahan K., Deltas C., Hosking A., Kmoch S., Rampoldi L., et al. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015;88:676–683. doi: 10.1038/ki.2015.28. - DOI - PubMed
-
- Bolar N.A., Golzio C., Živná M., Hayot G., Van Hemelrijk C., Schepers D., Vandeweyer G., Hoischen A., Huyghe J.R., Raes A., et al. Heterozygous Loss-of-Function SEC61A1 Mutations Cause Autosomal-Dominant Tubulo-Interstitial and Glomerulocystic Kidney Disease with Anemia. Am. J. Hum. Genet. 2016;99:174–187. doi: 10.1016/j.ajhg.2016.05.028. - DOI - PMC - PubMed
-
- Ayasreh N., Bullich G., Miquel R., Furlano M., Ruiz P., Lorente L., Valero O., García-González M.A., Arhda N., Garin I., et al. Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients With ADTKD-UMOD and ADTKD-MUC1. Am. J. Kidney Dis. 2018;72:411–418. doi: 10.1053/j.ajkd.2018.03.019. - DOI - PubMed
LinkOut - more resources
Research Materials
Miscellaneous