

Intermolecular electron transfer in radical SAM enzymes as a new paradigm for reductive activation
- PMID: 37460016
- PMCID: PMC10470005
- DOI: 10.1016/j.jbc.2023.105058
Intermolecular electron transfer in radical SAM enzymes as a new paradigm for reductive activation
Abstract
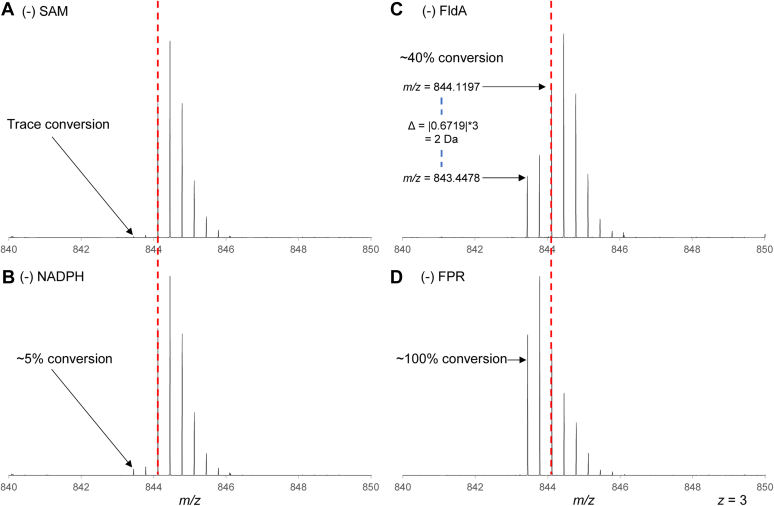
Radical S-adenosyl-L-methionine (rSAM) enzymes bind one or more Fe-S clusters and catalyze transformations that produce complex and structurally diverse natural products. One of the clusters, a 4Fe-4S cluster, binds and reductively cleaves SAM to generate the 5'-deoxyadenosyl radical, which initiates the catalytic cycle by H-atom transfer from the substrate. The role(s) of the additional auxiliary Fe-S clusters (ACs) remains largely enigmatic. The rSAM enzyme PapB catalyzes the formation of thioether cross-links between the β-carbon of an Asp and a Cys thiolate found in the PapA peptide. One of the two ACs in the protein binds to the substrate thiol where, upon formation of a thioether bond, one reducing equivalent is returned to the protein. However, for the next catalytic cycle to occur, the protein must undergo an electronic state isomerization, returning the electron to the SAM-binding cluster. Using a series of iron-sulfur cluster deletion mutants, our data support a model whereby the isomerization is an obligatorily intermolecular electron transfer event that can be mediated by redox active proteins or small molecules, likely via the second AC in PapB. Surprisingly, a mixture of FMN and NADPH is sufficient to support both the reductive and the isomerization steps. These findings lead to a new paradigm involving intermolecular electron transfer steps in the activation of rSAM enzymes that require multiple iron-sulfur clusters for turnover. The implications of these results for the biological activation of rSAM enzymes are discussed.
Keywords: S-adenosyl-L-methionine (SAM); electron transfer; enzymatic activation; enzyme mechanism; iron–sulfur protein; radical SAM.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest V. B. and K. A. S. E. have disclosed the results to the University of Utah, which holds patent interests in the findings.
Figures







Similar articles
-
Peptide Selenocysteine Substitutions Reveal Direct Substrate-Enzyme Interactions at Auxiliary Clusters in Radical S-Adenosyl-l-methionine Maturases.J Am Chem Soc. 2023 May 10;145(18):10167-10177. doi: 10.1021/jacs.3c00831. Epub 2023 Apr 27. J Am Chem Soc. 2023. PMID: 37104670 Free PMC article.
-
Mechanism of Radical Initiation in the Radical S-Adenosyl-l-methionine Superfamily.Acc Chem Res. 2018 Nov 20;51(11):2611-2619. doi: 10.1021/acs.accounts.8b00356. Epub 2018 Oct 15. Acc Chem Res. 2018. PMID: 30346729 Free PMC article. Review.
-
Trapping and Electron Paramagnetic Resonance Characterization of the 5'dAdo• Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate.ACS Cent Sci. 2019 Nov 27;5(11):1777-1785. doi: 10.1021/acscentsci.9b00706. Epub 2019 Sep 25. ACS Cent Sci. 2019. PMID: 31807679 Free PMC article.
-
Auxiliary iron-sulfur cofactors in radical SAM enzymes.Biochim Biophys Acta. 2015 Jun;1853(6):1316-34. doi: 10.1016/j.bbamcr.2015.01.002. Epub 2015 Jan 15. Biochim Biophys Acta. 2015. PMID: 25597998 Review.
-
Characterization of auxiliary iron-sulfur clusters in a radical S-adenosylmethionine enzyme PqqE from Methylobacterium extorquens AM1.FEBS Open Bio. 2017 Oct 18;7(12):1864-1879. doi: 10.1002/2211-5463.12314. eCollection 2017 Dec. FEBS Open Bio. 2017. PMID: 29226074 Free PMC article.
Cited by
-
Diverse thioether macrocyclized peptides through a radical SAM maturase.Proc Natl Acad Sci U S A. 2025 Aug 26;122(34):e2512563122. doi: 10.1073/pnas.2512563122. Epub 2025 Aug 21. Proc Natl Acad Sci U S A. 2025. PMID: 40838878 Free PMC article.
-
Insights into the Mechanism of Installation of 5-Carboxymethylaminomethyl Uridine Hypermodification by tRNA-Modifying Enzymes MnmE and MnmG.J Am Chem Soc. 2023 Dec 13;145(49):26947-26961. doi: 10.1021/jacs.3c10182. Epub 2023 Dec 5. J Am Chem Soc. 2023. PMID: 38050996 Free PMC article.
-
Biochemical and genetic studies define the functions of methylthiotransferases in methanogenic and methanotrophic archaea.Front Microbiol. 2023 Nov 23;14:1304671. doi: 10.3389/fmicb.2023.1304671. eCollection 2023. Front Microbiol. 2023. PMID: 38075885 Free PMC article.
-
Initiation, Propagation, and Termination in the Chemistry of Radical SAM Enzymes.Biochemistry. 2024 Dec 17;63(24):3161-3183. doi: 10.1021/acs.biochem.4c00518. Epub 2024 Dec 3. Biochemistry. 2024. PMID: 39626071 Free PMC article. Review.
-
A [2.1.0]-Fused Bicyclic Intermediate Is Produced during the Biosynthesis of Oxetane Nucleosides.J Am Chem Soc. 2025 Jul 23;147(29):25224-25232. doi: 10.1021/jacs.5c01831. Epub 2025 Jul 11. J Am Chem Soc. 2025. PMID: 40643284 Free PMC article.
References
-
- Oberg N., Precord T.W., Mitchell D.A., Gerlt J.A. RadicalSAM.org: a resource to interpret sequence-function space and discover new radical SAM enzyme chemistry. ACS Bio. Med. Chem. Au. 2022;2:22–35. - PMC - PubMed
-
- Sofia H.J., Chen G., Hetzler B.G., Reyes-Spindola J.F., Miller N.E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucl. Acids Res. 2001;29:1097. - PMC - PubMed
-
- Krebs C., Broderick W.E., Henshaw T.F., Broderick J.B., Huynh B.H. Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating anzyme: a Mössbauer spectroscopic study. J. Am. Chem. Soc. 2002;124:912–913. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
