

Inhibition of Abelson Tyrosine-Protein Kinase 2 Suppresses the Development of Alcohol-Associated Liver Disease by Decreasing PPARgamma Expression
- PMID: 37460041
- PMCID: PMC10520367
- DOI: 10.1016/j.jcmgh.2023.07.006
Inhibition of Abelson Tyrosine-Protein Kinase 2 Suppresses the Development of Alcohol-Associated Liver Disease by Decreasing PPARgamma Expression
Abstract
Background & aims: Alcohol-associated liver disease (ALD) represents a spectrum of alcohol use-related liver diseases. Outside of alcohol abstinence, there are currently no Food and Drug Administration-approved treatments for advanced ALD, necessitating a greater understanding of ALD pathogenesis and potential molecular targets for therapeutic intervention. The ABL-family proteins, including ABL1 and ABL2, are non-receptor tyrosine kinases that participate in a diverse set of cellular functions. We investigated the role of the ABL kinases in alcohol-associated liver disease.
Methods: We used samples from patients with ALD compared with healthy controls to elucidate a clinical phenotype. We established strains of liver-specific Abl1 and Abl2 knockout mice and subjected them to the National Institute on Alcohol Abuse and Alcoholism acute-on-chronic alcohol feeding regimen. Murine samples were subjected to RNA sequencing, AST, Oil Red O staining, H&E staining, Western blotting, and quantitative polymerase chain reaction to assess phenotypic changes after alcohol feeding. In vitro modeling in HepG2 cells as well as primary hepatocytes from C57BL6/J mice was used to establish this mechanistic link of ALD pathogenesis.
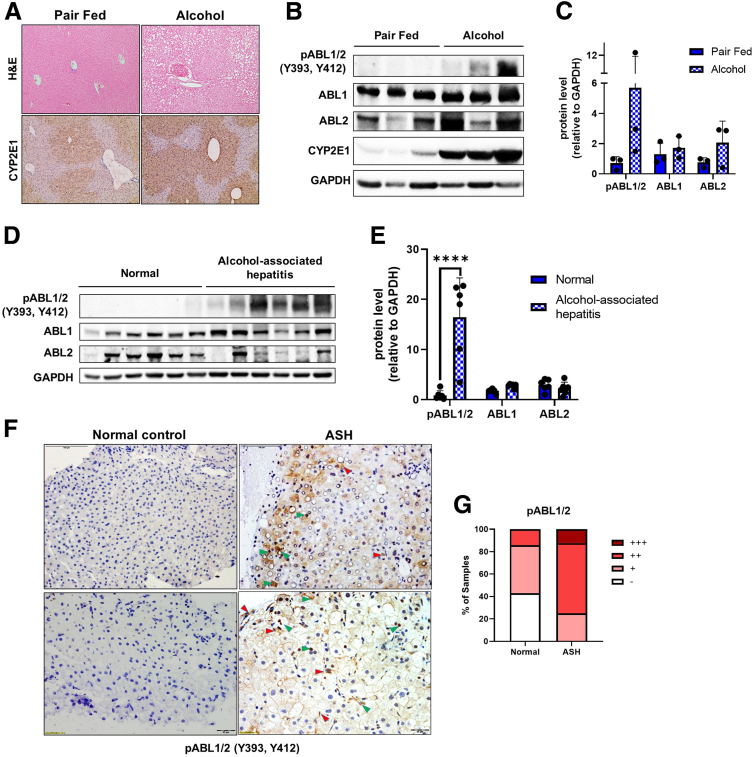
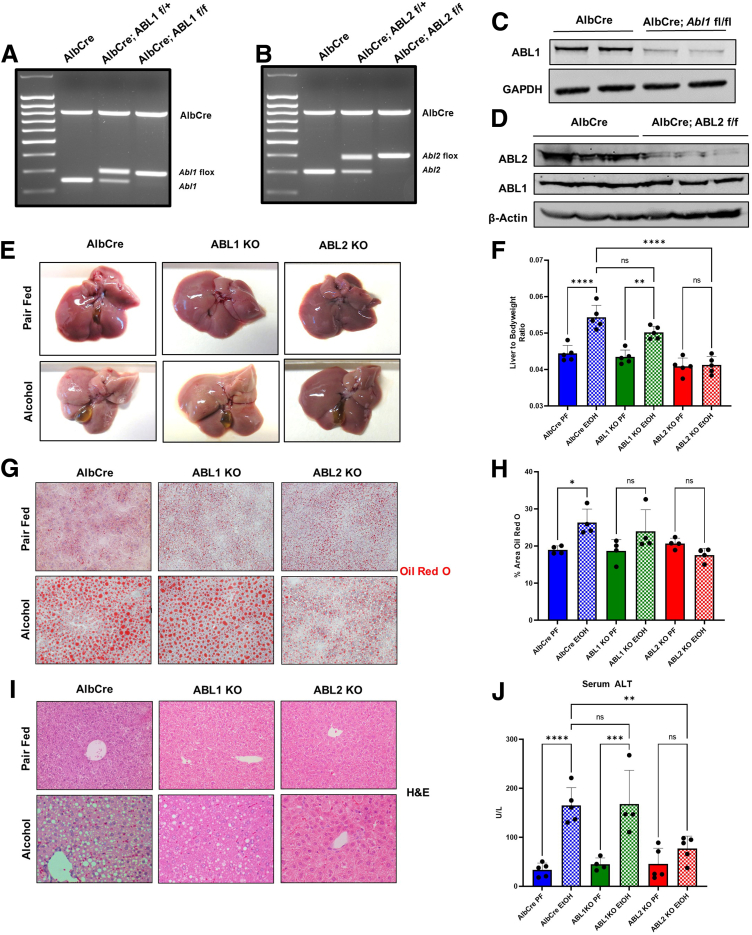
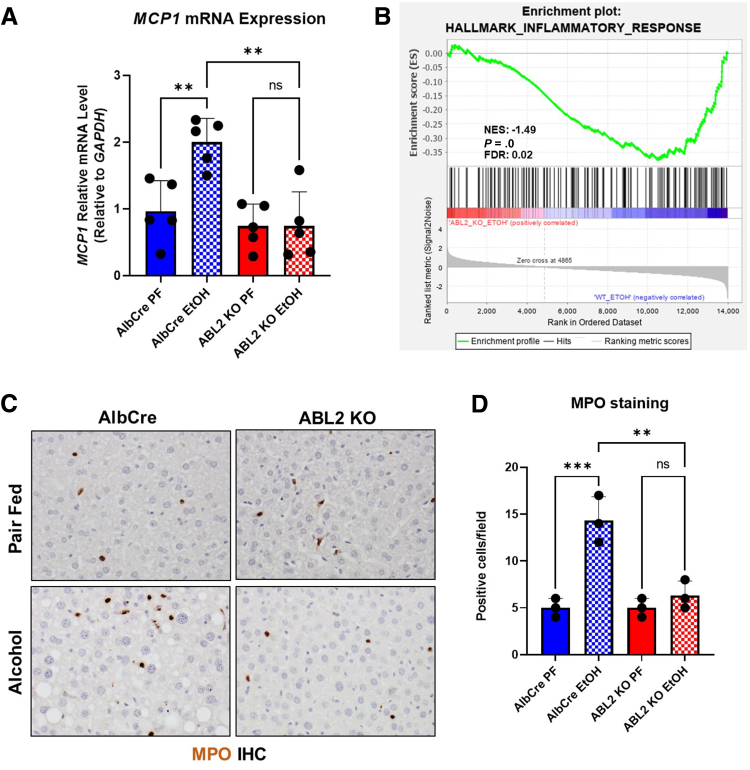
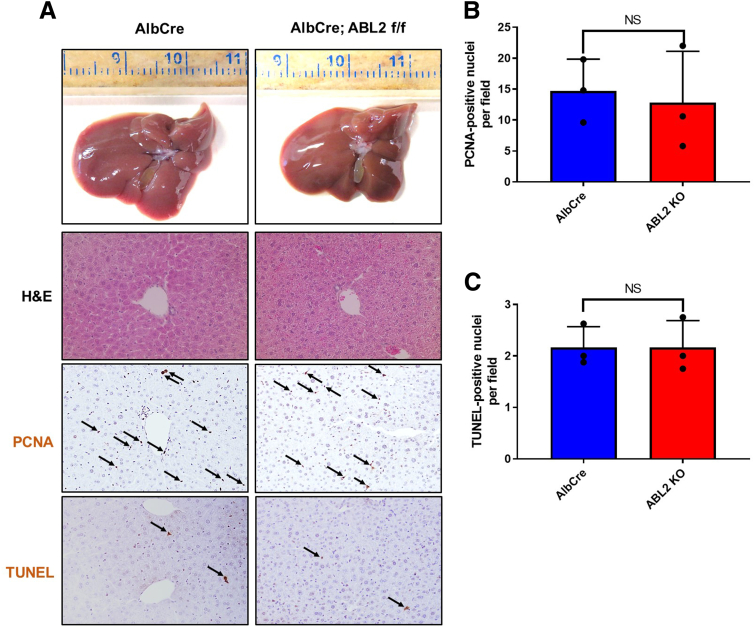
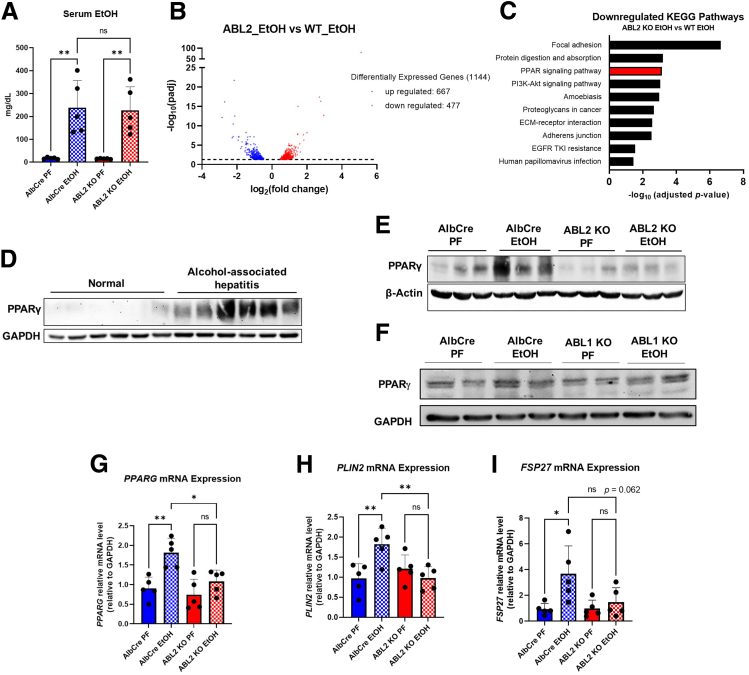
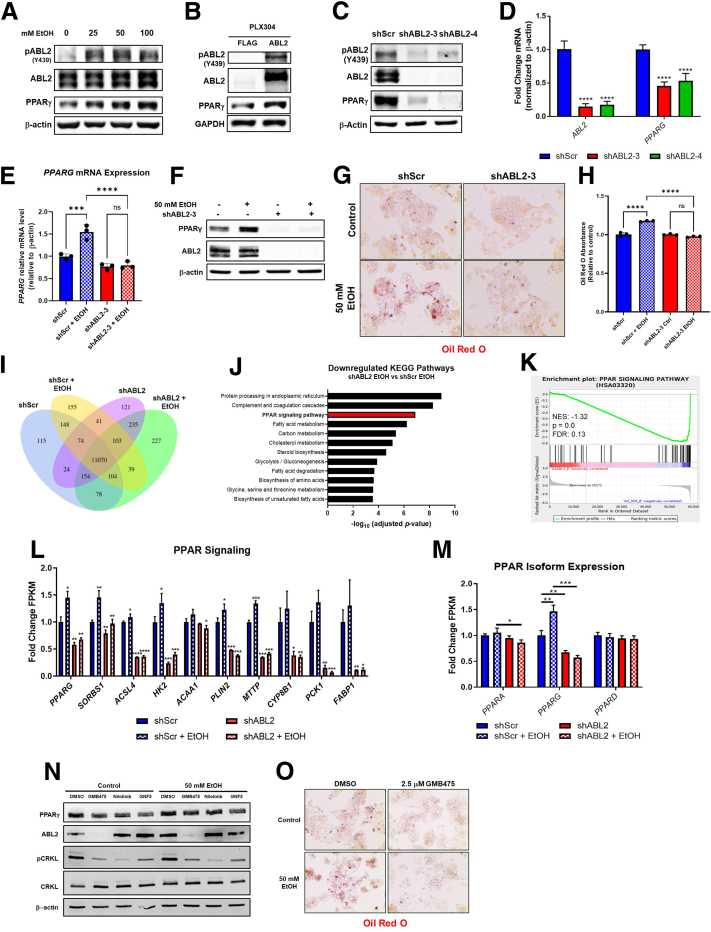
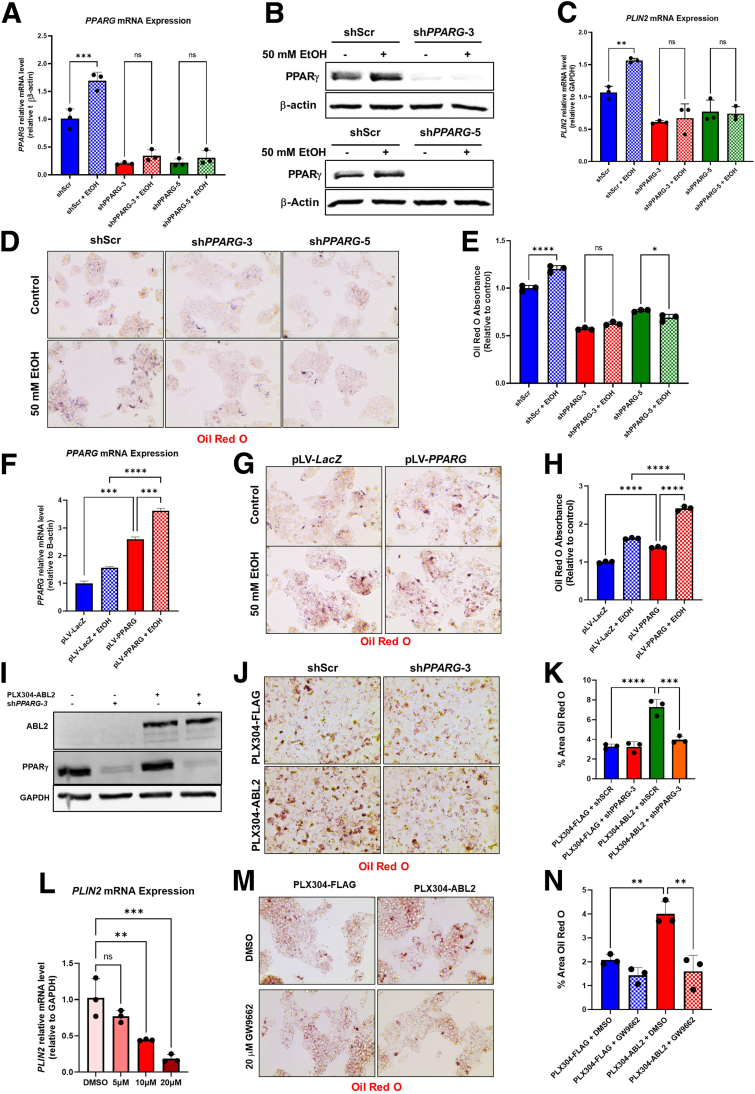
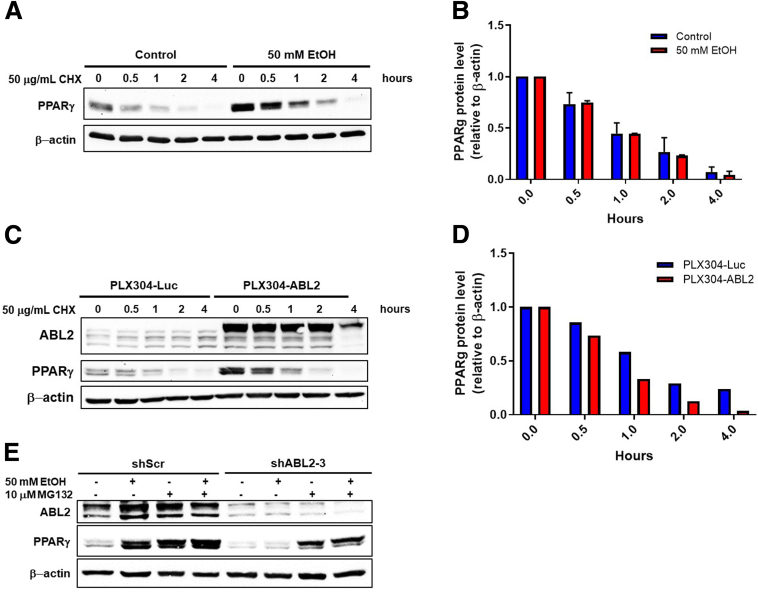
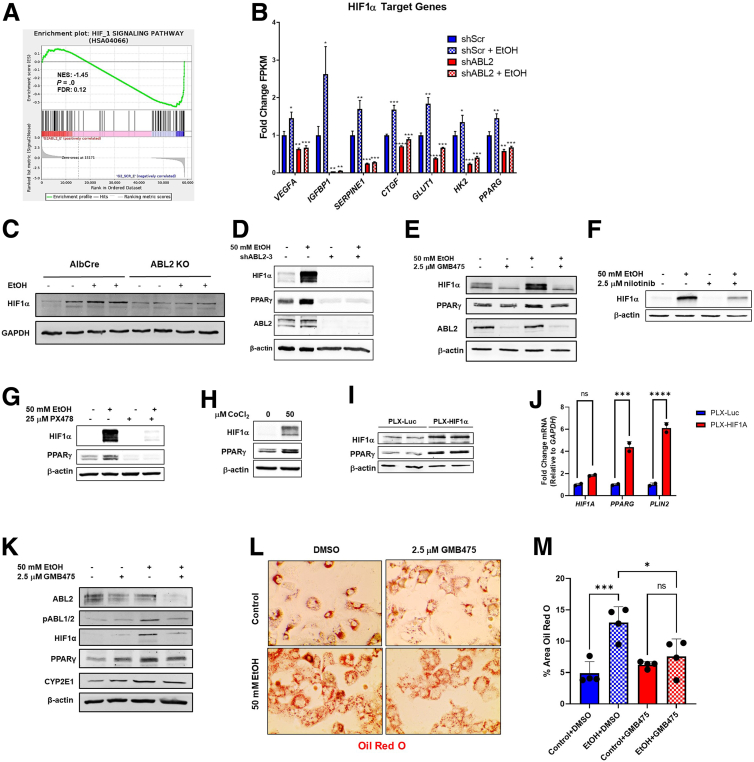
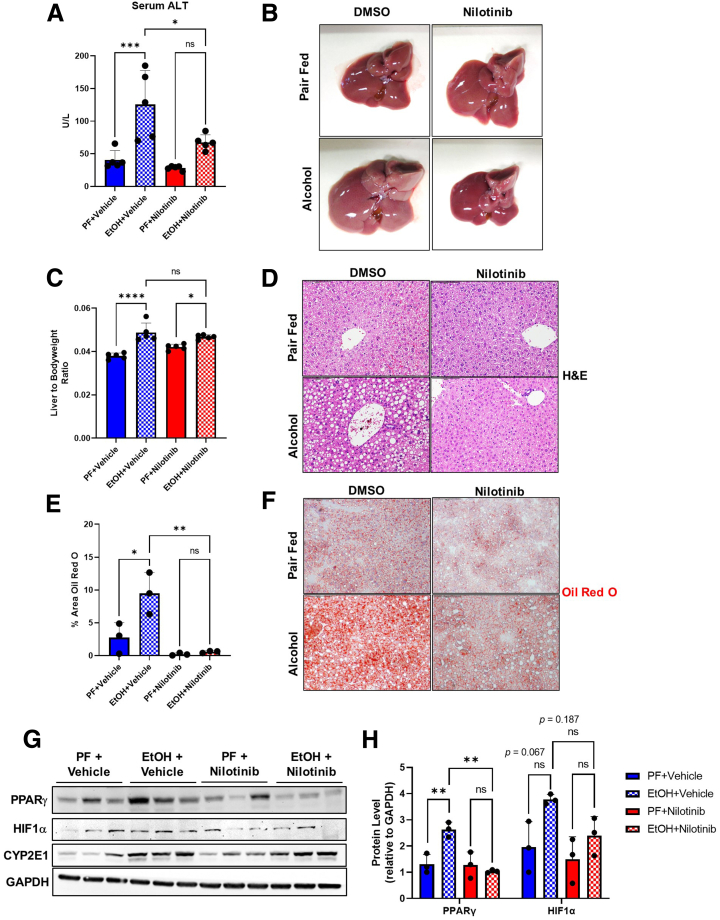
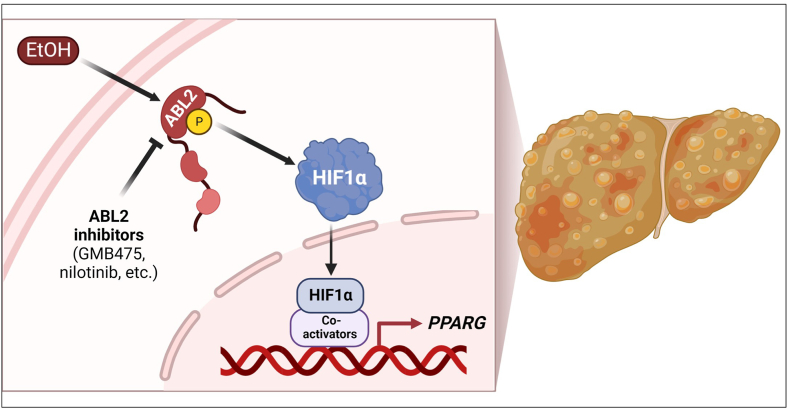
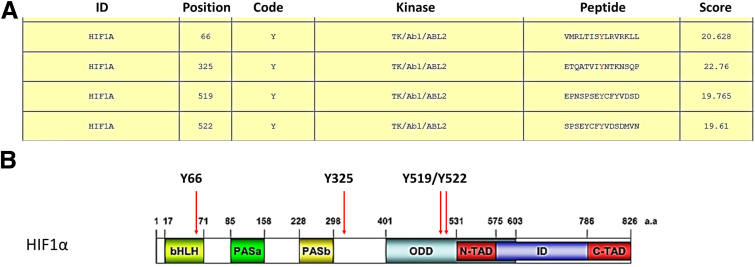
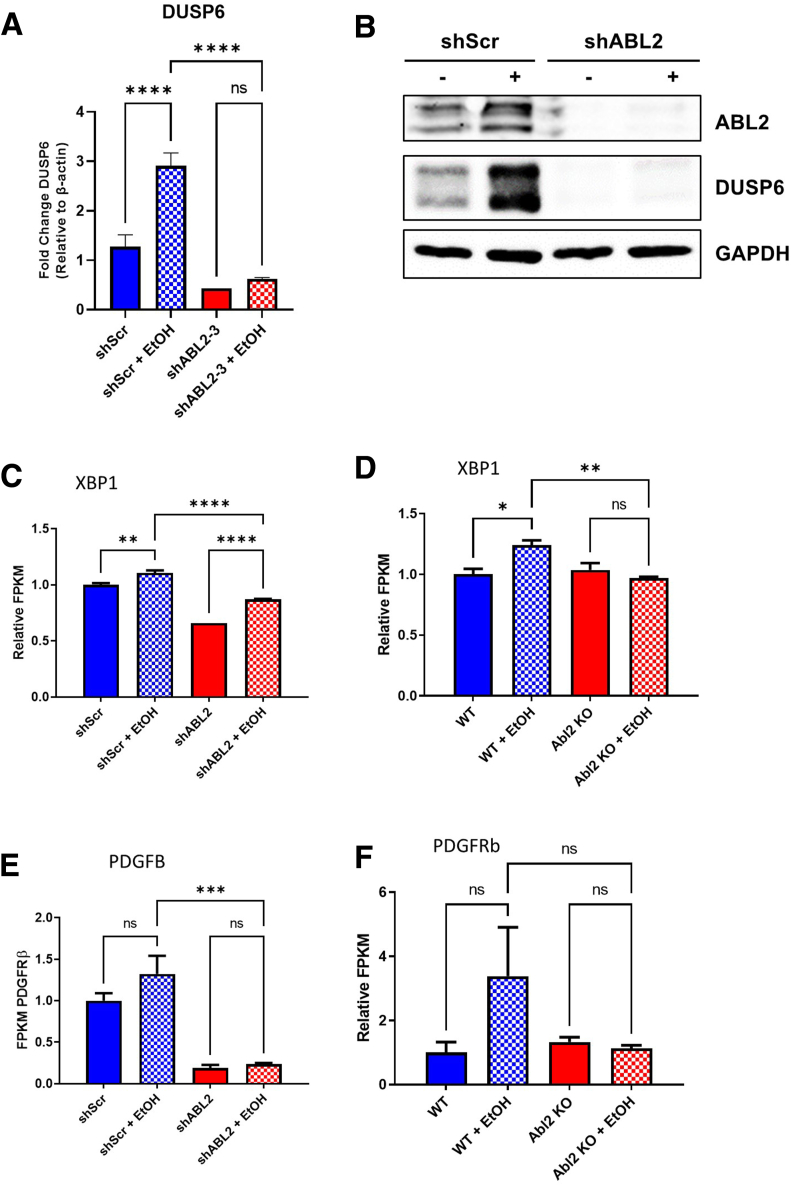
Results: We demonstrate that the ABL kinases are highly activated in ALD patient liver samples as well as in liver tissues from mice subjected to an alcohol feeding regimen. We found that the liver-specific knockout of Abl2, but not Abl1, attenuated alcohol-induced steatosis, liver injury, and inflammation. Subsequent RNA sequencing and gene set enrichment analyses of mouse liver tissues revealed that relative to wild-type alcohol-fed mice, Abl2 knockout alcohol-fed mice exhibited numerous pathway changes, including significantly decreased peroxisome proliferator activated receptor (PPAR) signaling. Further examination revealed that PPARγ, a previously identified regulator of ALD pathogenesis, was induced upon alcohol feeding in wild-type mice, but not in Abl2 knockout mice. In vitro analyses revealed that shRNA-mediated knockdown of ABL2 abolished the alcohol-induced accumulation of PPARγ as well as subsequent lipid accumulation. Conversely, forced overexpression of ABL2 resulted in increased PPARγ protein expression. Furthermore, we demonstrated that the regulation of hypoxia inducible factor 1 subunit alpha (HIF1α) by ABL2 is required for alcohol-induced PPARγ expression. Furthermore, treatment with ABL kinase inhibitors attenuated alcohol-induced PPARγ expression, lipid droplet formation, and liver injury.
Conclusions: On the basis of our current evidence, we propose that alcohol-induced ABL2 activation promotes ALD through increasing HIF1α and the subsequent PPARγ expression, and ABL2 inhibition may serve as a promising target for the treatment of ALD.
Keywords: ABL2; Alcohol-Associated Liver Disease; HIF1α; Nilotinib; PPARγ.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Lieber C.S. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
