

This is a preprint.
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank
- PMID: 37461573
- PMCID: PMC10350147
- DOI: 10.1101/2023.06.29.23291992
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank
Update in
-
Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank.Cell Genom. 2024 Jul 10;4(7):100602. doi: 10.1016/j.xgen.2024.100602. Epub 2024 Jun 28. Cell Genom. 2024. PMID: 38944039 Free PMC article.
Abstract
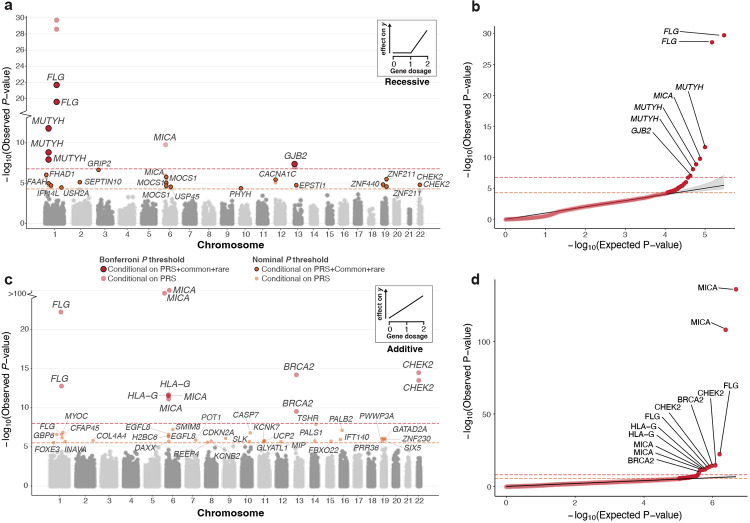
Exome-sequencing association studies have successfully linked rare protein-coding variation to risk of thousands of diseases. However, the relationship between rare deleterious compound heterozygous (CH) variation and their phenotypic impact has not been fully investigated. Here, we leverage advances in statistical phasing to accurately phase rare variants (MAF ~ 0.001%) in exome sequencing data from 175,587 UK Biobank (UKBB) participants, which we then systematically annotate to identify putatively deleterious CH coding variation. We show that 6.5% of individuals carry such damaging variants in the CH state, with 90% of variants occurring at MAF < 0.34%. Using a logistic mixed model framework, systematically accounting for relatedness, polygenic risk, nearby common variants, and rare variant burden, we investigate recessive effects in common complex diseases. We find six exome-wide significant () and 17 nominally significant () gene-trait associations. Among these, only four would have been identified without accounting for CH variation in the gene. We further incorporate age-at-diagnosis information from primary care electronic health records, to show that genetic phase influences lifetime risk of disease across 20 gene-trait combinations (FDR < 5%). Using a permutation approach, we find evidence for genetic phase contributing to disease susceptibility for a collection of gene-trait pairs, including FLG-asthma () and USH2A-visual impairment (). Taken together, we demonstrate the utility of phasing large-scale genetic sequencing cohorts for robust identification of the phenome-wide consequences of compound heterozygosity.
Figures




References
-
- Nelson M. R., Tipney H., Painter J. L., et al. The support of human genetic evidence for approved drug indications. en. Nature Genetics 47, 856–860 (Aug. 2015). - PubMed
-
- Plenge R. M., Scolnick E. M. & Altshuler D. Validating therapeutic targets through human genetics. en. Nature Reviews Drug Discovery 12. Number: 8 Publisher: Nature Publishing Group, 581–594 (Aug. 2013). - PubMed
-
- Tobert J. A. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. en. Nature Reviews Drug Discovery 2. Number: 7 Publisher: Nature Publishing Group, 517–526 (July 2003). - PubMed
-
- Do R. Q., Vogel R. A. & Schwartz G. G. PCSK9 Inhibitors: potential in cardiovascular therapeutics. eng. Current Cardiology Reports 15, 345 (Mar. 2013). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous