

Parameter Estimation and Species Tree Rooting Using ALE and GeneRax
- PMID: 37463417
- PMCID: PMC10373948
- DOI: 10.1093/gbe/evad134
Parameter Estimation and Species Tree Rooting Using ALE and GeneRax
Abstract
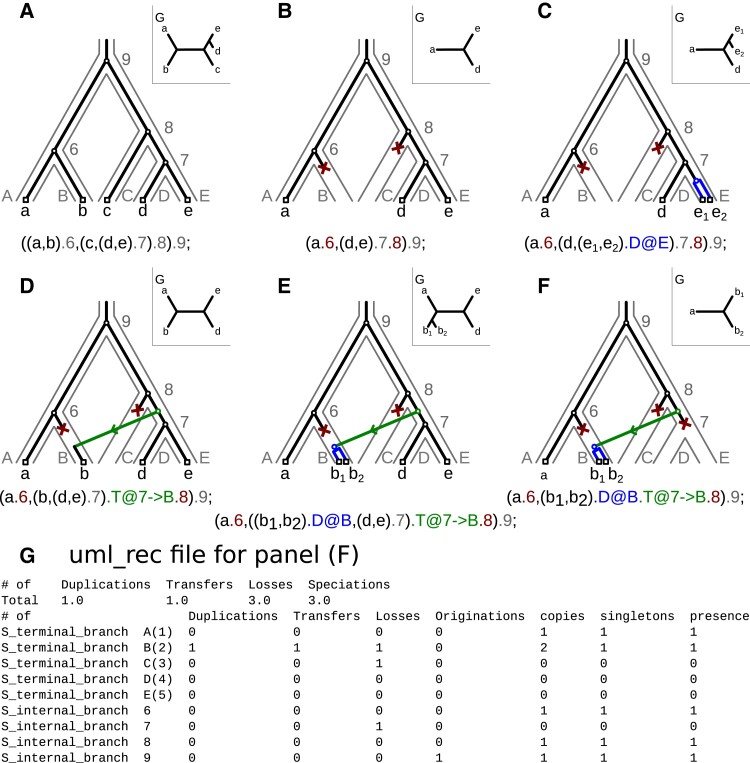
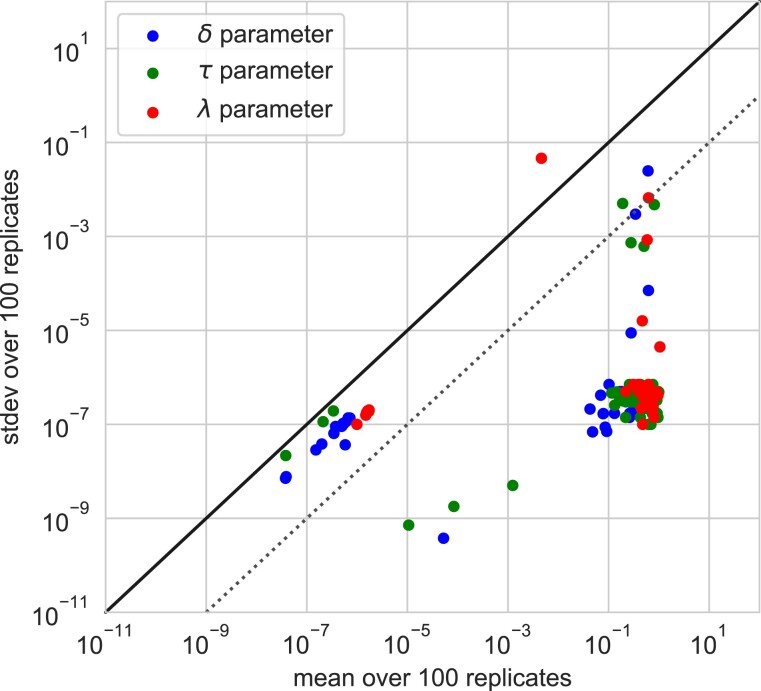
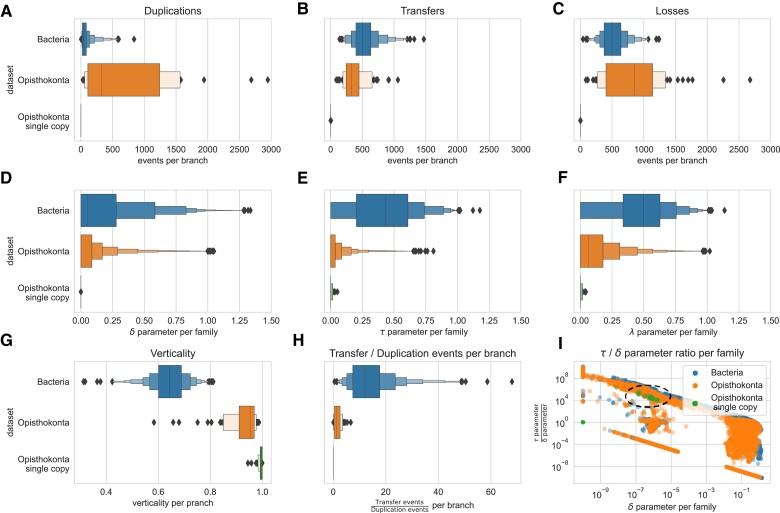
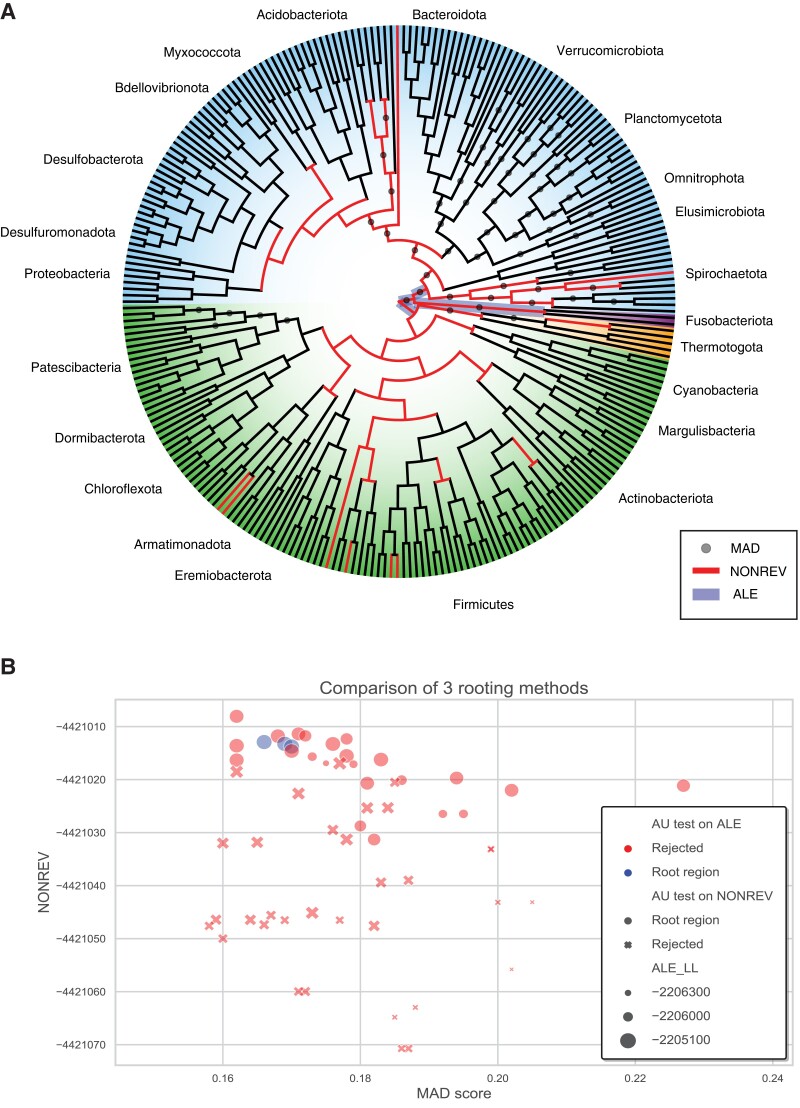
ALE and GeneRax are tools for probabilistic gene tree-species tree reconciliation. Based on a common underlying statistical model of how gene trees evolve along species trees, these methods rely on gene vs. species tree discordance to infer gene duplication, transfer, and loss events, map gene family origins, and root species trees. Published analyses have used these methods to root species trees of Archaea, Bacteria, and several eukaryotic groups, as well as to infer ancestral gene repertoires. However, it was recently suggested that reconciliation-based estimates of duplication and transfer events using the ALE/GeneRax model were unreliable, with potential implications for species tree rooting. Here, we assess these criticisms and find that the methods are accurate when applied to simulated data and in generally good agreement with alternative methodological approaches on empirical data. In particular, ALE recovers variation in gene duplication and transfer frequencies across lineages that is consistent with the known biology of studied clades. In plants and opisthokonts, ALE recovers the consensus species tree root; in Bacteria-where there is less certainty about the root position-ALE agrees with alternative approaches on the most likely root region. Overall, ALE and related approaches are promising tools for studying genome evolution.
Keywords: comparative genomics; gene tree–species tree reconciliation; microbial evolution; phylogenetics.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
Similar articles
-
Realistic Gene Transfer to Gene Duplication Ratios Identify Different Roots in the Bacterial Phylogeny Using a Tree Reconciliation Method.Life (Basel). 2022 Jul 4;12(7):995. doi: 10.3390/life12070995. Life (Basel). 2022. PMID: 35888084 Free PMC article.
-
GeneRax: A Tool for Species-Tree-Aware Maximum Likelihood-Based Gene Family Tree Inference under Gene Duplication, Transfer, and Loss.Mol Biol Evol. 2020 Sep 1;37(9):2763-2774. doi: 10.1093/molbev/msaa141. Mol Biol Evol. 2020. PMID: 32502238 Free PMC article.
-
On the impact of uncertain gene tree rooting on duplication-transfer-loss reconciliation.BMC Bioinformatics. 2018 Aug 13;19(Suppl 9):290. doi: 10.1186/s12859-018-2269-0. BMC Bioinformatics. 2018. PMID: 30367593 Free PMC article.
-
Phylogenetic reconciliation: making the most of genomes to understand microbial ecology and evolution.ISME J. 2024 Jan 8;18(1):wrae129. doi: 10.1093/ismejo/wrae129. ISME J. 2024. PMID: 39001714 Free PMC article. Review.
-
The inference of gene trees with species trees.Syst Biol. 2015 Jan;64(1):e42-62. doi: 10.1093/sysbio/syu048. Epub 2014 Jul 28. Syst Biol. 2015. PMID: 25070970 Free PMC article. Review.
Cited by
-
The origin, evolution, and molecular diversity of the chemokine system.Life Sci Alliance. 2024 Jan 16;7(3):e202302471. doi: 10.26508/lsa.202302471. Print 2024 Mar. Life Sci Alliance. 2024. PMID: 38228369 Free PMC article.
-
Genomics of soil depth niche partitioning in the Thaumarchaeota family Gagatemarchaeaceae.Nat Commun. 2023 Nov 11;14(1):7305. doi: 10.1038/s41467-023-43196-0. Nat Commun. 2023. PMID: 37951938 Free PMC article.
-
Microbial Diversity and Open Questions about the Deep Tree of Life.Genome Biol Evol. 2024 Apr 2;16(4):evae053. doi: 10.1093/gbe/evae053. Genome Biol Evol. 2024. PMID: 38620144 Free PMC article.
-
The emerging view on the origin and early evolution of eukaryotic cells.Nature. 2024 Sep;633(8029):295-305. doi: 10.1038/s41586-024-07677-6. Epub 2024 Sep 11. Nature. 2024. PMID: 39261613 Review.
References
-
- Battistuzzi FU, Blair Hedges S. 2009. A major clade of prokaryotes with ancient adaptations to life on land. Mol Biol Evol. 26(2):335–343. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
