

Distinct shared and compartment-enriched oncogenic networks drive primary versus metastatic breast cancer
- PMID: 37463901
- PMCID: PMC10354065
- DOI: 10.1038/s41467-023-39935-y
Distinct shared and compartment-enriched oncogenic networks drive primary versus metastatic breast cancer
Abstract
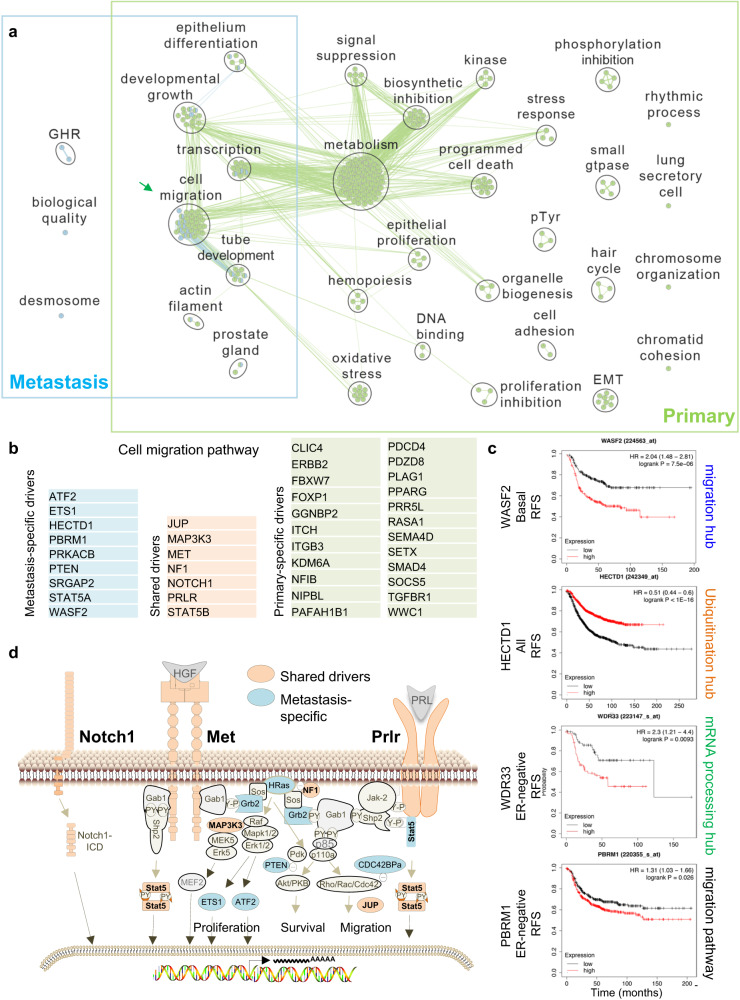
Metastatic breast-cancer is a major cause of death in women worldwide, yet the relationship between oncogenic drivers that promote metastatic versus primary cancer is still contentious. To elucidate this relationship in treatment-naive animals, we hereby describe mammary-specific transposon-mutagenesis screens in female mice together with loss-of-function Rb, which is frequently inactivated in breast-cancer. We report gene-centric common insertion-sites (gCIS) that are enriched in primary-tumors, in metastases or shared by both compartments. Shared-gCIS comprise a major MET-RAS network, whereas metastasis-gCIS form three additional hubs: Rho-signaling, Ubiquitination and RNA-processing. Pathway analysis of four clinical cohorts with paired primary-tumors and metastases reveals similar organization in human breast-cancer with subtype-specific shared-drivers (e.g. RB1-loss, TP53-loss, high MET, RAS, ER), primary-enriched (EGFR, TGFβ and STAT3) and metastasis-enriched (RHO, PI3K) oncogenic signaling. Inhibitors of RB1-deficiency or MET plus RHO-signaling cooperate to block cell migration and drive tumor cell-death. Thus, targeting shared- and metastasis- but not primary-enriched derivers offers a rational avenue to prevent metastatic breast-cancer.
© 2023. The Author(s).
Conflict of interest statement
C.M.P. is an equity stockholder and consultant of BioClassifier LLC; C.M.P. is also listed as an inventor on patent applications for the Breast PAM50 Subtyping assay. There is no direct relationship between these PAM50 patents and the Intellectual Property and content of this study. The other authors declare no conflict of interest.
Figures







References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
