

Chromatin regulators in the TBX1 network confer risk for conotruncal heart defects in 22q11.2DS
- PMID: 37463940
- PMCID: PMC10354062
- DOI: 10.1038/s41525-023-00363-y
Chromatin regulators in the TBX1 network confer risk for conotruncal heart defects in 22q11.2DS
Abstract
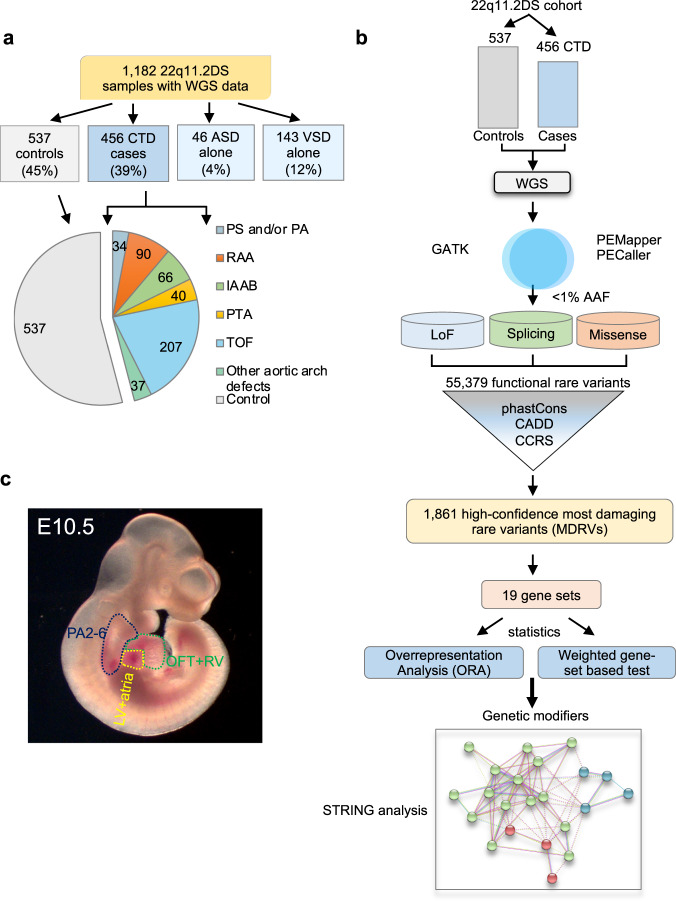
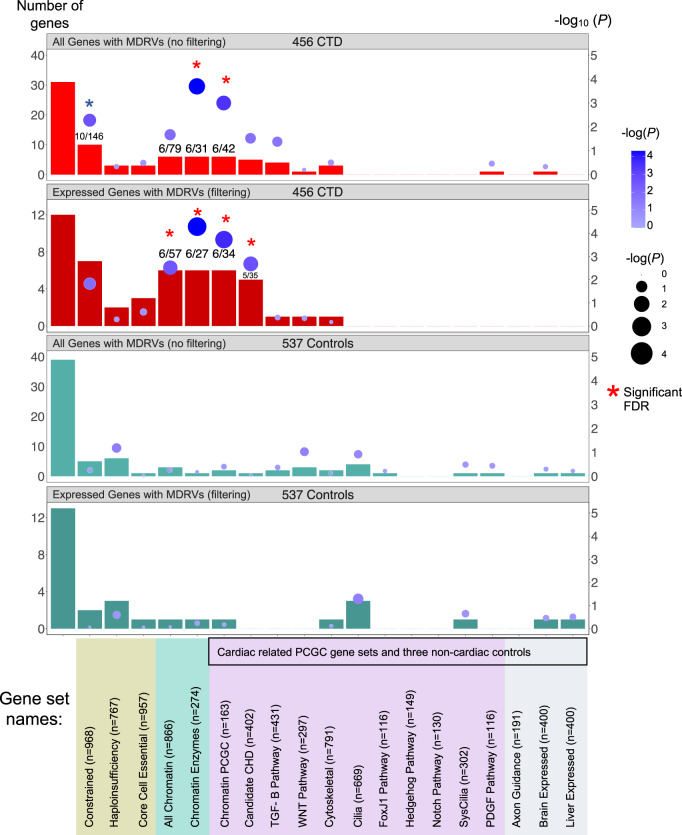
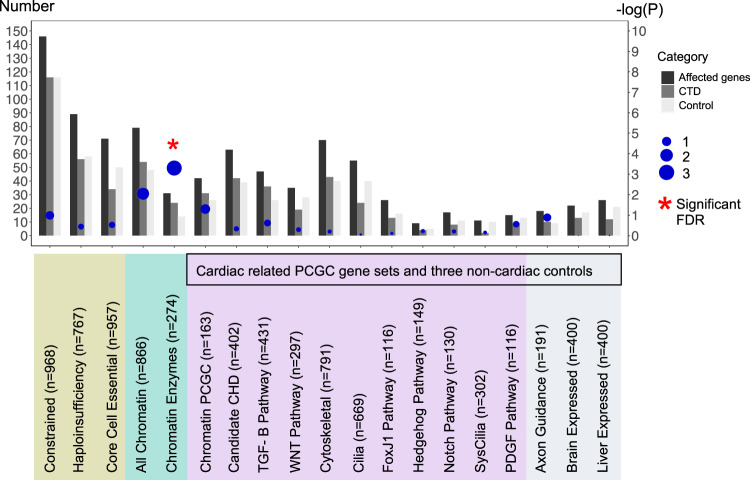
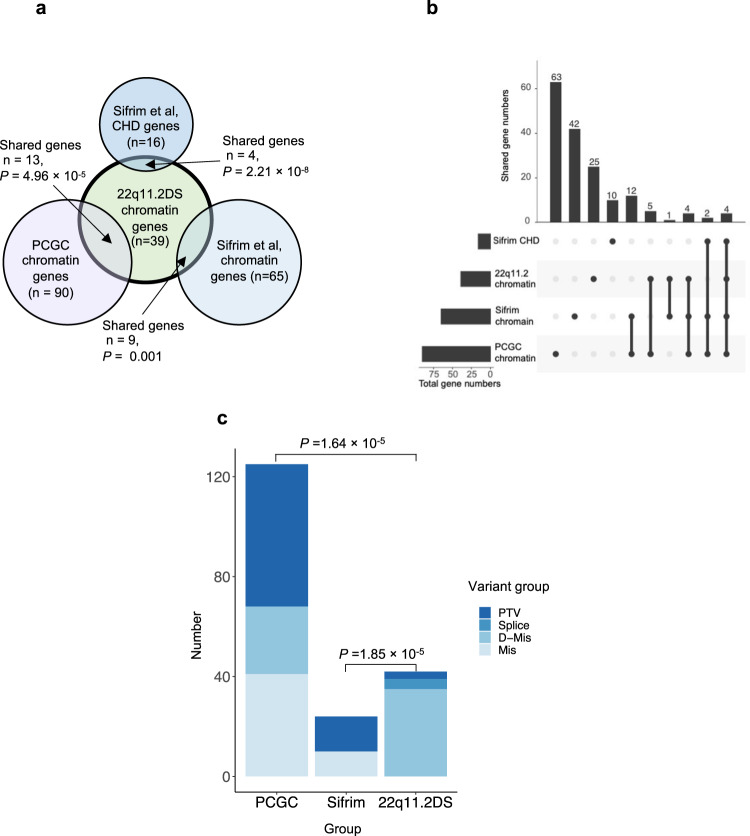
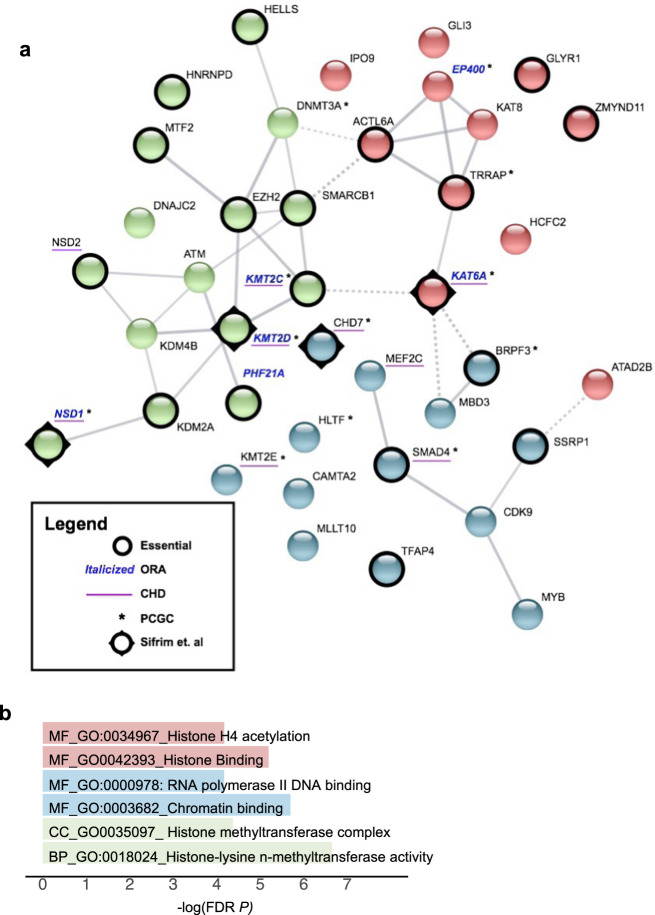
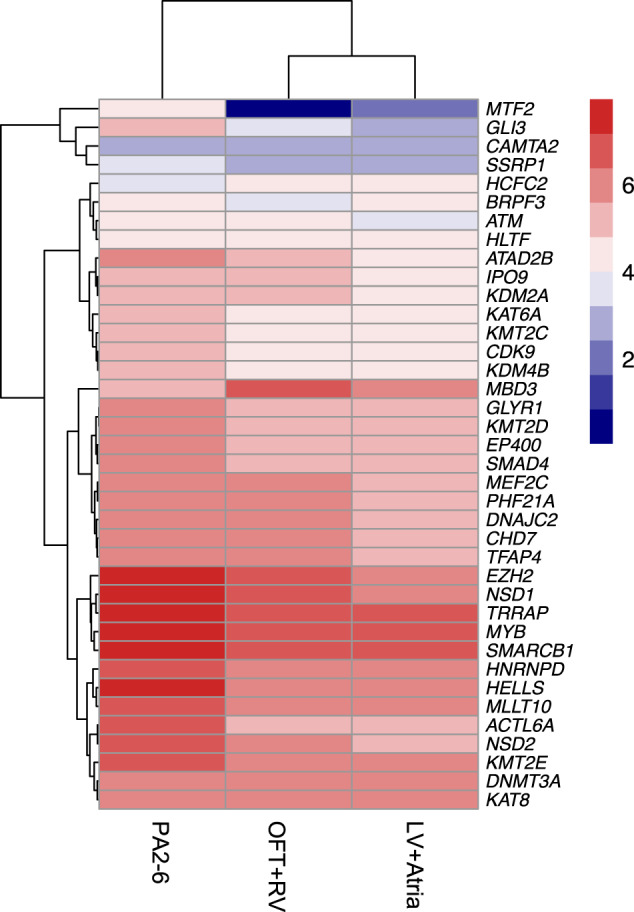
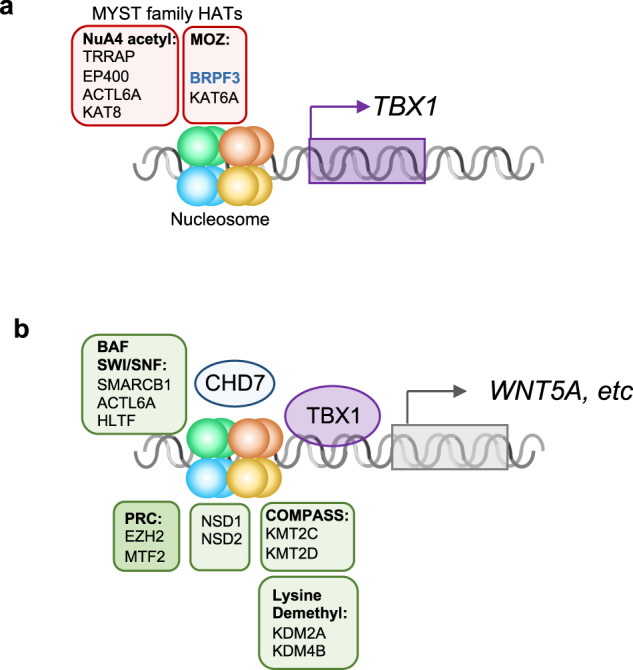
Congenital heart disease (CHD) affecting the conotruncal region of the heart, occurs in 40-50% of patients with 22q11.2 deletion syndrome (22q11.2DS). This syndrome is a rare disorder with relative genetic homogeneity that can facilitate identification of genetic modifiers. Haploinsufficiency of TBX1, encoding a T-box transcription factor, is one of the main genes responsible for the etiology of the syndrome. We suggest that genetic modifiers of conotruncal defects in patients with 22q11.2DS may be in the TBX1 gene network. To identify genetic modifiers, we analyzed rare, predicted damaging variants in whole genome sequence of 456 cases with conotruncal defects and 537 controls, with 22q11.2DS. We then performed gene set approaches and identified chromatin regulatory genes as modifiers. Chromatin genes with recurrent damaging variants include EP400, KAT6A, KMT2C, KMT2D, NSD1, CHD7 and PHF21A. In total, we identified 37 chromatin regulatory genes, that may increase risk for conotruncal heart defects in 8.5% of 22q11.2DS cases. Many of these genes were identified as risk factors for sporadic CHD in the general population. These genes are co-expressed in cardiac progenitor cells with TBX1, suggesting that they may be in the same genetic network. The genes KAT6A, KMT2C, CHD7 and EZH2, have been previously shown to genetically interact with TBX1 in mouse models. Our findings indicate that disturbance of chromatin regulatory genes impact the TBX1 gene network serving as genetic modifiers of 22q11.2DS and sporadic CHD, suggesting that there are some shared mechanisms involving the TBX1 gene network in the etiology of CHD.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing financial interests. There are competing non-financial interests for JASV and MJO. JASV has served as a consultant for NoBias Therapeutics, Inc for the design of a medication trial for individuals with 22q11DS (unrelated to the content of this work). MJO reports a research grant from Takeda Pharmaceuticals outside the scope of the current work. The remaining authors declare no competing interests.
Figures
Comment in
-
22q11.2 Deletion Syndrome: Influence of Parental Origin on Clinical Heterogeneity.Genes (Basel). 2024 Apr 21;15(4):518. doi: 10.3390/genes15040518. Genes (Basel). 2024. PMID: 38674452 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
