

SCN1A-deficient excitatory neuronal networks display mutation-specific phenotypes
- PMID: 37467479
- PMCID: PMC10689919
- DOI: 10.1093/brain/awad245
SCN1A-deficient excitatory neuronal networks display mutation-specific phenotypes
Abstract
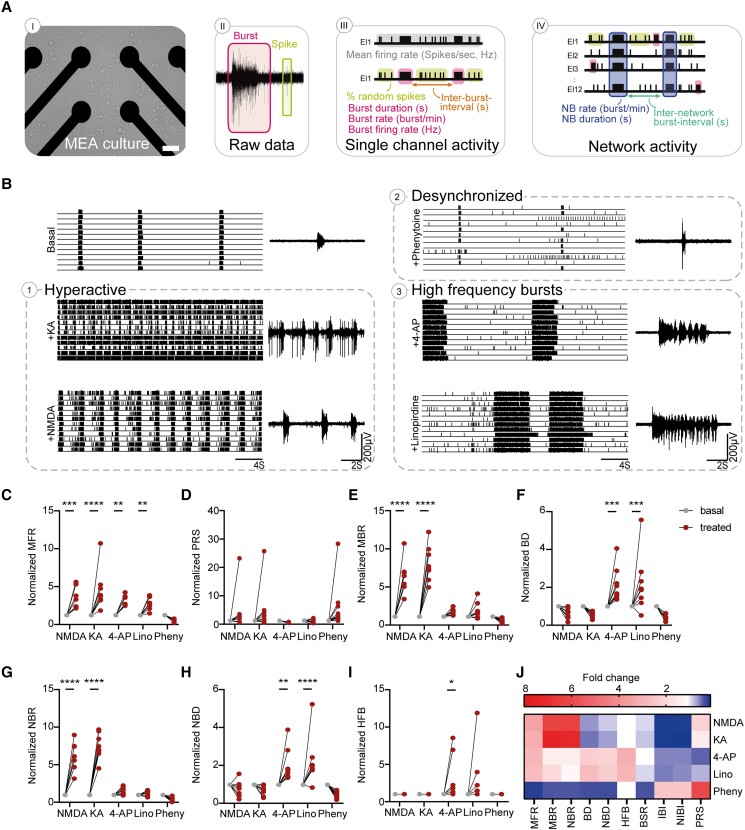
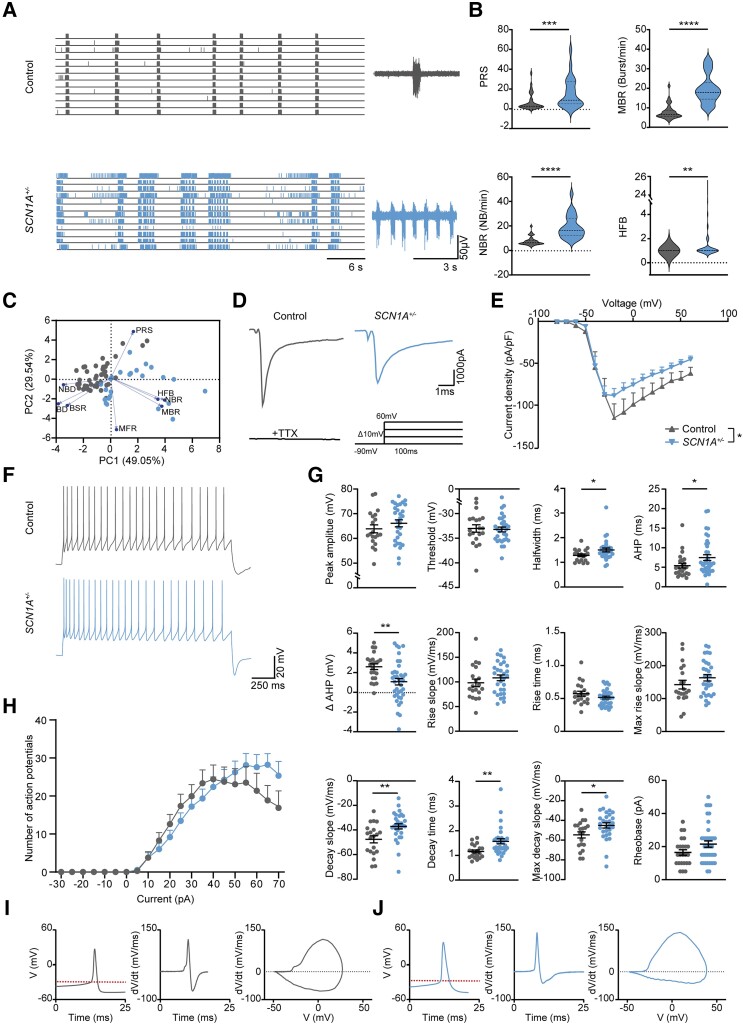
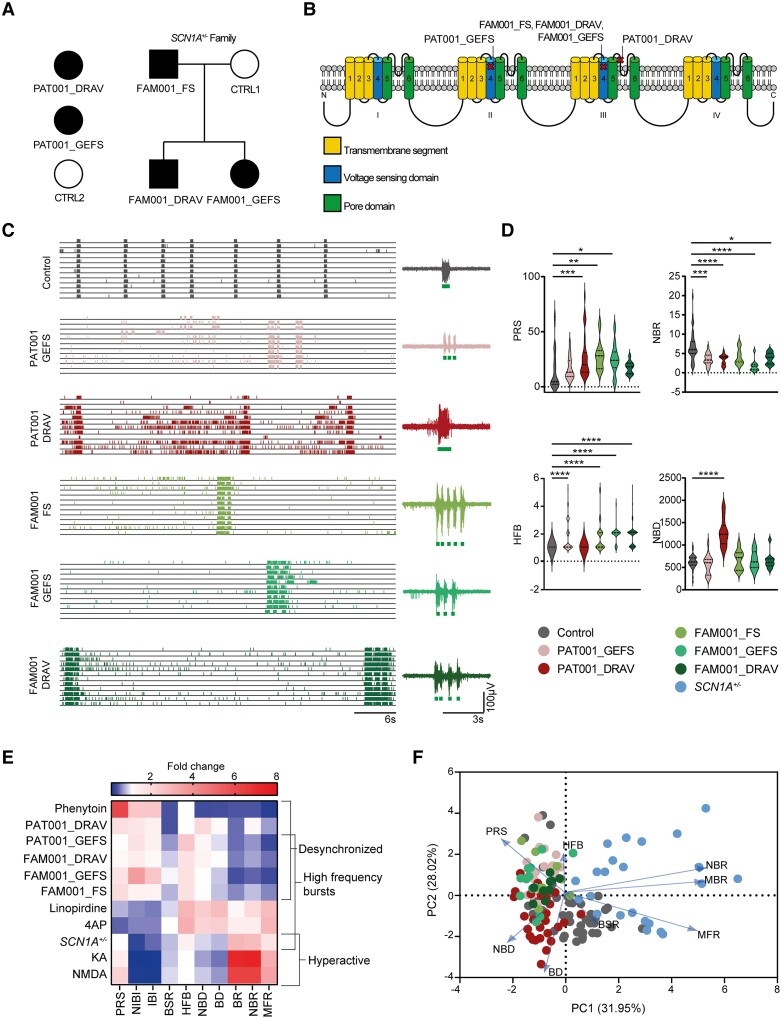
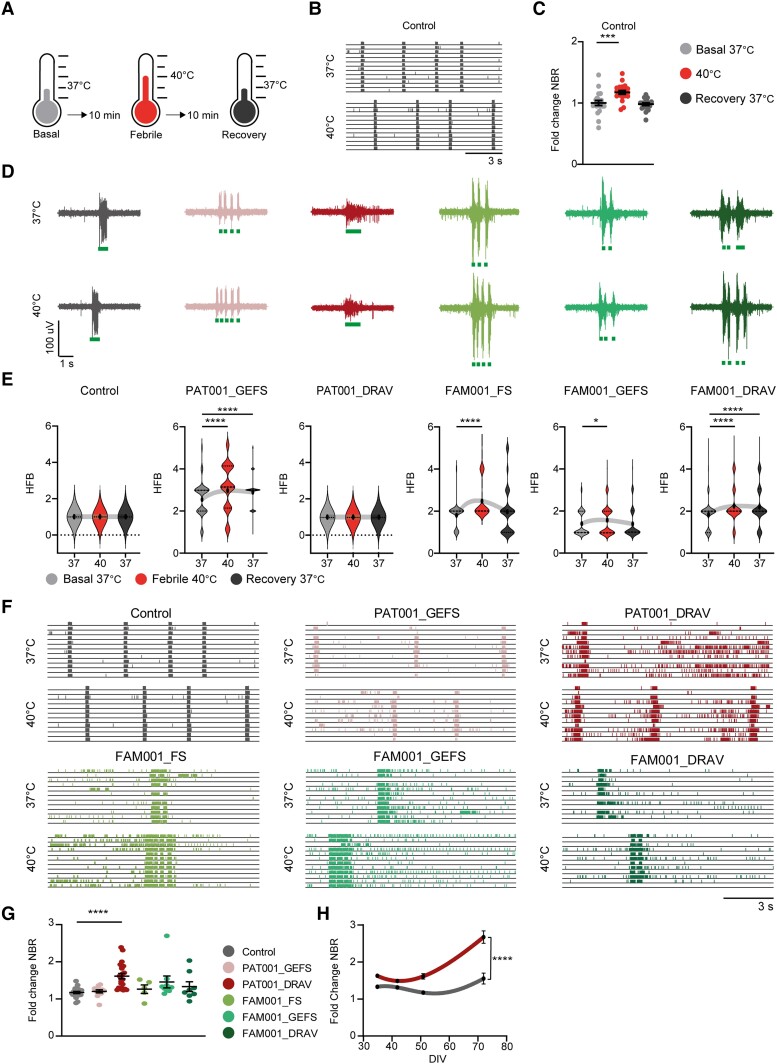
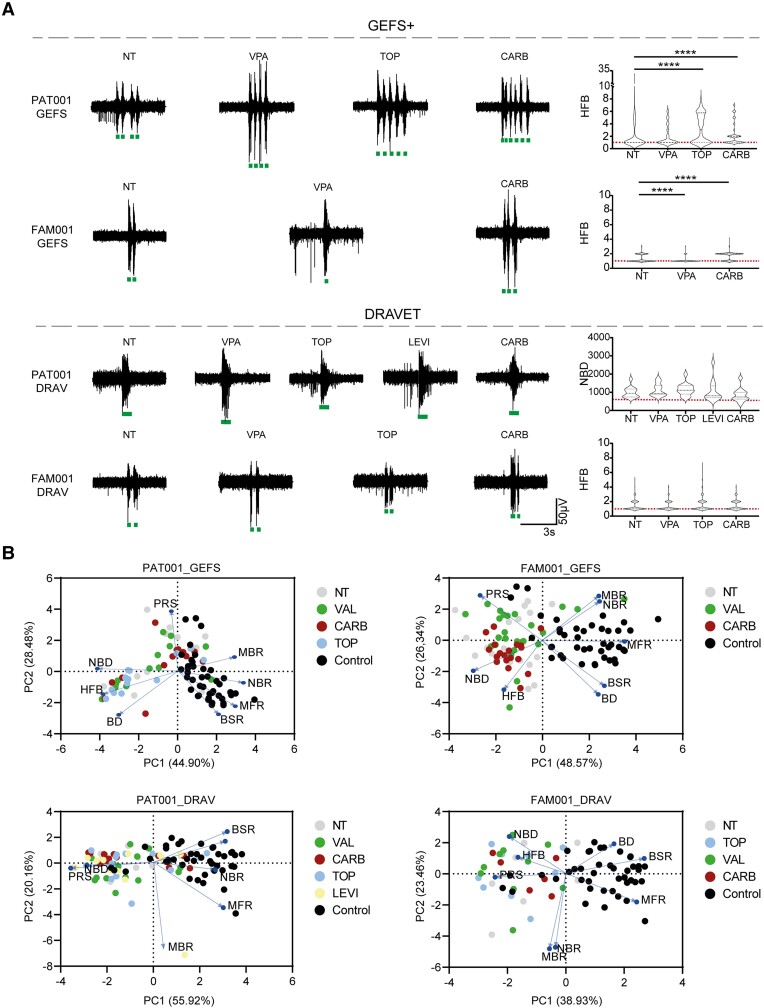
Dravet syndrome is a severe epileptic encephalopathy, characterized by (febrile) seizures, behavioural problems and developmental delay. Eighty per cent of patients with Dravet syndrome have a mutation in SCN1A, encoding Nav1.1. Milder clinical phenotypes, such as GEFS+ (generalized epilepsy with febrile seizures plus), can also arise from SCN1A mutations. Predicting the clinical phenotypic outcome based on the type of mutation remains challenging, even when the same mutation is inherited within one family. This clinical and genetic heterogeneity adds to the difficulties of predicting disease progression and tailoring the prescription of anti-seizure medication. Understanding the neuropathology of different SCN1A mutations may help to predict the expected clinical phenotypes and inform the selection of best-fit treatments. Initially, the loss of Na+-current in inhibitory neurons was recognized specifically to result in disinhibition and consequently seizure generation. However, the extent to which excitatory neurons contribute to the pathophysiology is currently debated and might depend on the patient clinical phenotype or the specific SCN1A mutation. To examine the genotype-phenotype correlations of SCN1A mutations in relation to excitatory neurons, we investigated a panel of patient-derived excitatory neuronal networks differentiated on multi-electrode arrays. We included patients with different clinical phenotypes, harbouring various SCN1A mutations, along with a family in which the same mutation led to febrile seizures, GEFS+ or Dravet syndrome. We hitherto describe a previously unidentified functional excitatory neuronal network phenotype in the context of epilepsy, which corresponds to seizurogenic network prediction patterns elicited by proconvulsive compounds. We found that excitatory neuronal networks were affected differently, depending on the type of SCN1A mutation, but did not segregate according to clinical severity. Specifically, loss-of-function mutations could be distinguished from missense mutations, and mutations in the pore domain could be distinguished from mutations in the voltage sensing domain. Furthermore, all patients showed aggravated neuronal network responses at febrile temperatures compared with controls. Finally, retrospective drug screening revealed that anti-seizure medication affected GEFS+ patient- but not Dravet patient-derived neuronal networks in a patient-specific and clinically relevant manner. In conclusion, our results indicate a mutation-specific excitatory neuronal network phenotype, which recapitulates the foremost clinically relevant features, providing future opportunities for precision therapies.
Keywords: Dravet syndrome; MEA; epilepsy; hiPSC.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
References
-
- Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52:3–9. - PubMed
-
- Deprez L, Jansen A, De Jonghe P. Genetics of epilepsy syndromes starting in the first year of life. Neurology. 2009;72:273–281. - PubMed
-
- Wolff M, Cassé-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. 2006;47:45–48. - PubMed
-
- Jansen FE, Sadleir LG, Harkin LA, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology. 2006;67:2224–2226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
