

Mutant APC reshapes Wnt signaling plasma membrane nanodomains by altering cholesterol levels via oncogenic β-catenin
- PMID: 37468468
- PMCID: PMC10356786
- DOI: 10.1038/s41467-023-39640-w
Mutant APC reshapes Wnt signaling plasma membrane nanodomains by altering cholesterol levels via oncogenic β-catenin
Abstract
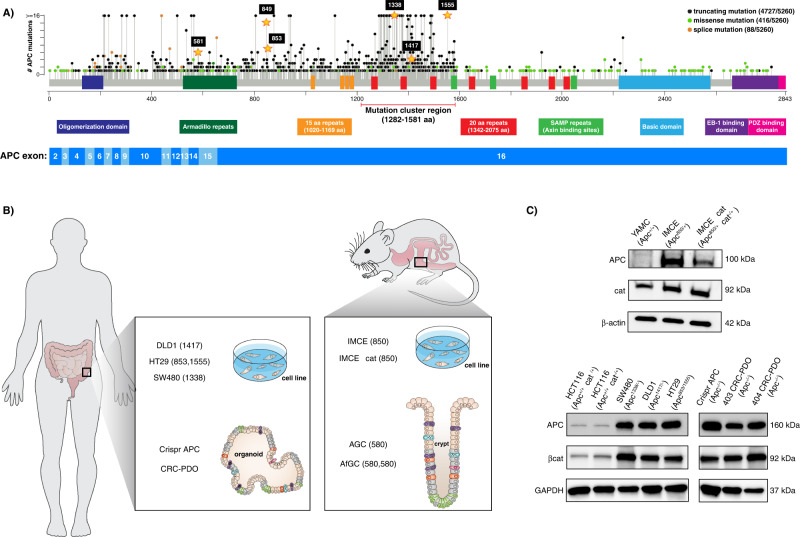
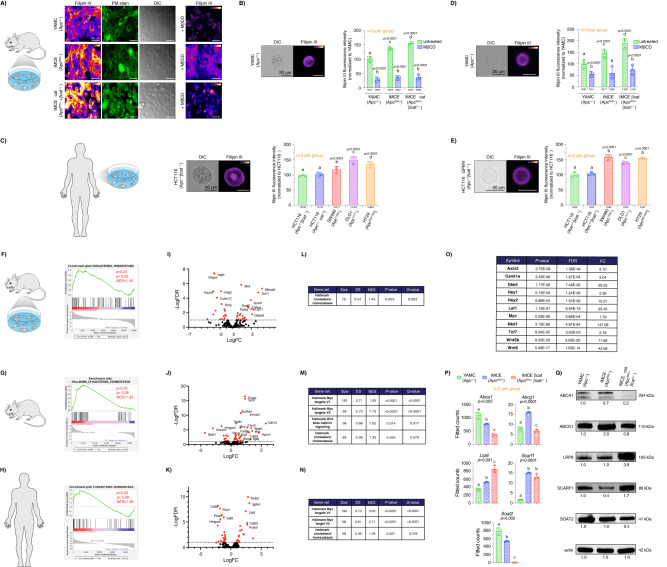
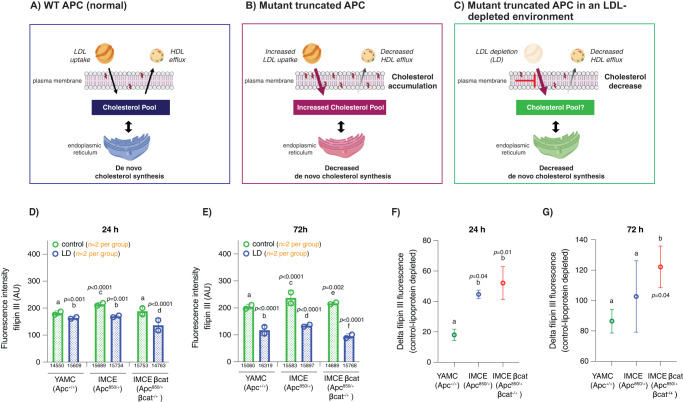
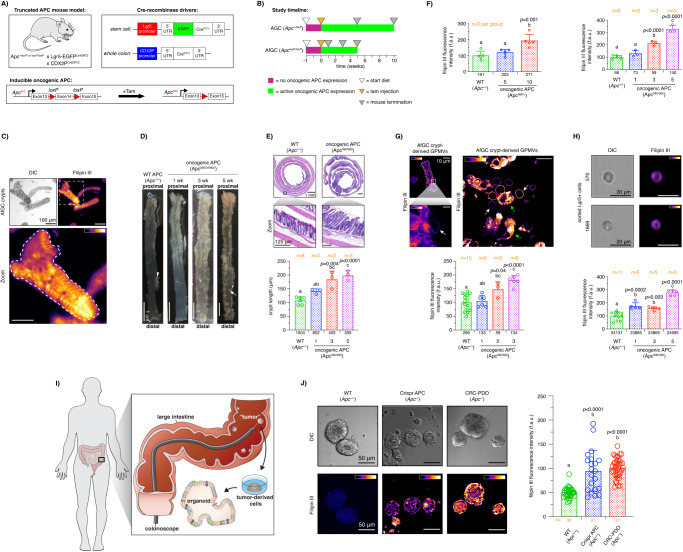
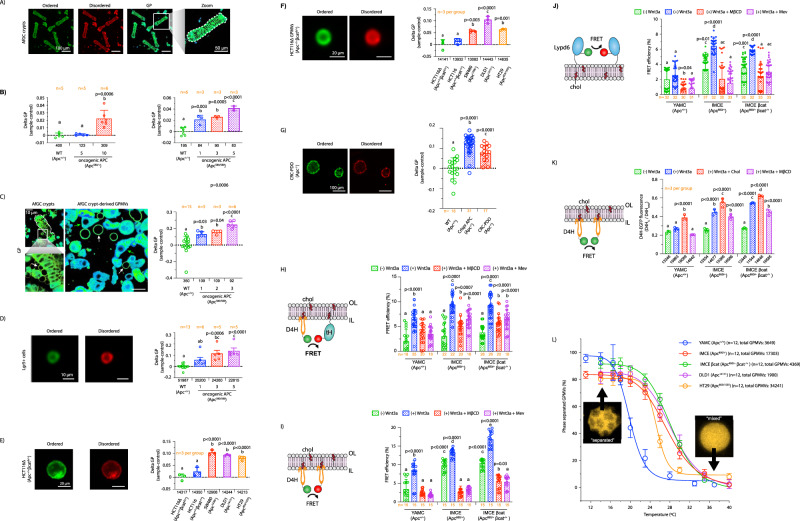
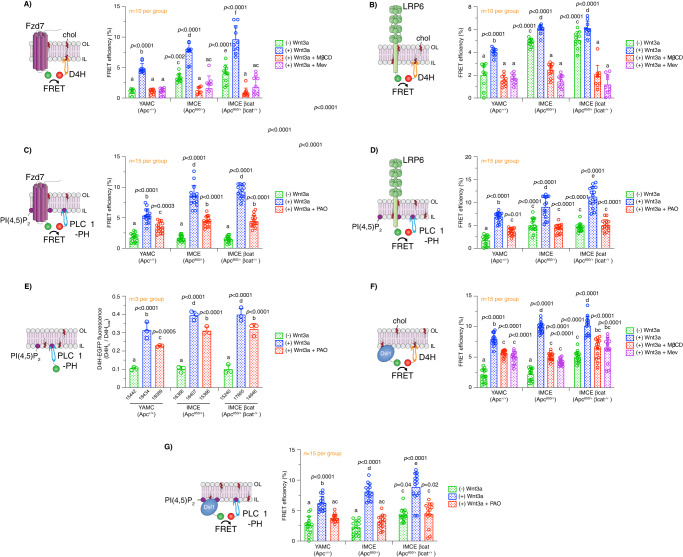
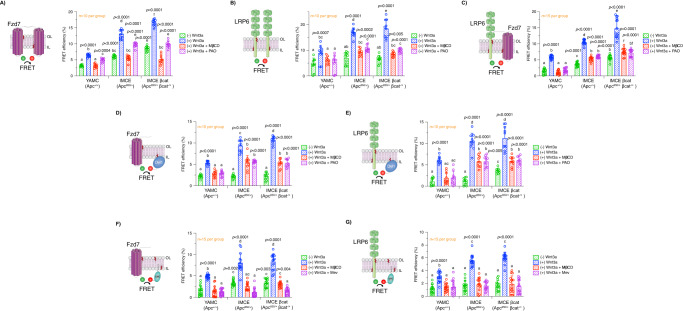
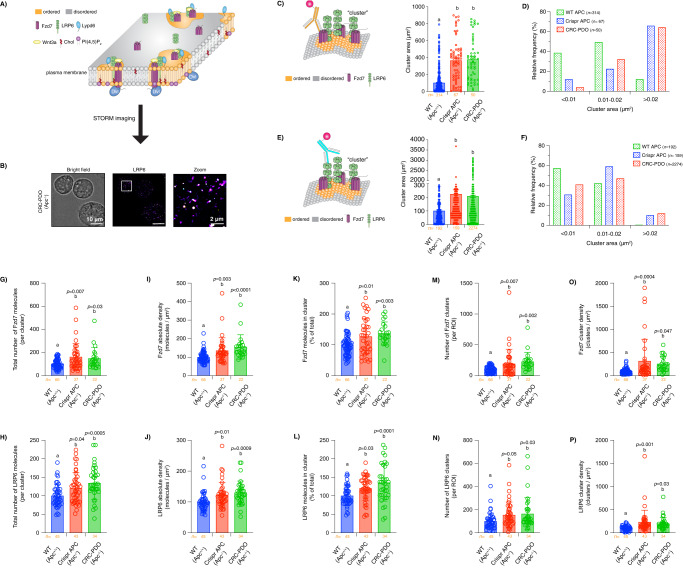
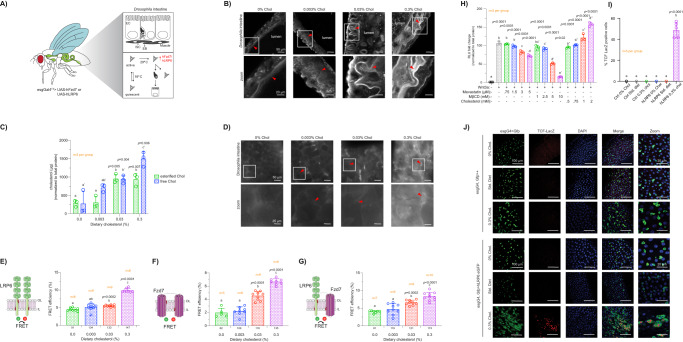
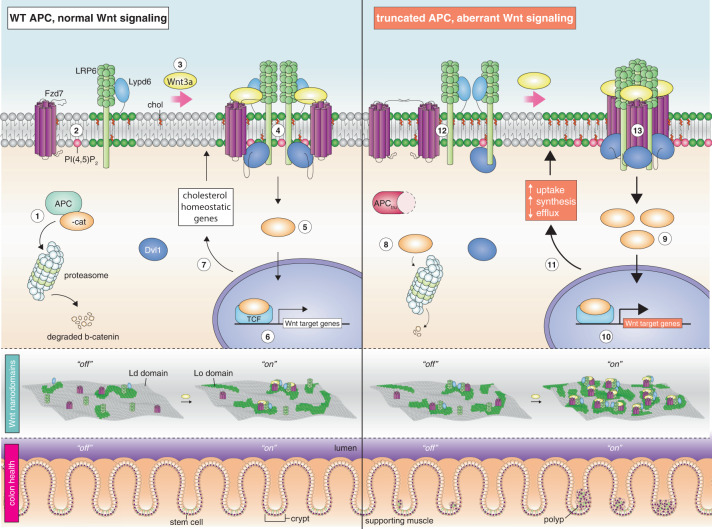
Although the role of the Wnt pathway in colon carcinogenesis has been described previously, it has been recently demonstrated that Wnt signaling originates from highly dynamic nano-assemblies at the plasma membrane. However, little is known regarding the role of oncogenic APC in reshaping Wnt nanodomains. This is noteworthy, because oncogenic APC does not act autonomously and requires activation of Wnt effectors upstream of APC to drive aberrant Wnt signaling. Here, we demonstrate the role of oncogenic APC in increasing plasma membrane free cholesterol and rigidity, thereby modulating Wnt signaling hubs. This results in an overactivation of Wnt signaling in the colon. Finally, using the Drosophila sterol auxotroph model, we demonstrate the unique ability of exogenous free cholesterol to disrupt plasma membrane homeostasis and drive Wnt signaling in a wildtype APC background. Collectively, these findings provide a link between oncogenic APC, loss of plasma membrane homeostasis and CRC development.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
