

An atlas of healthy and injured cell states and niches in the human kidney
- PMID: 37468583
- PMCID: PMC10356613
- DOI: 10.1038/s41586-023-05769-3
An atlas of healthy and injured cell states and niches in the human kidney
Abstract
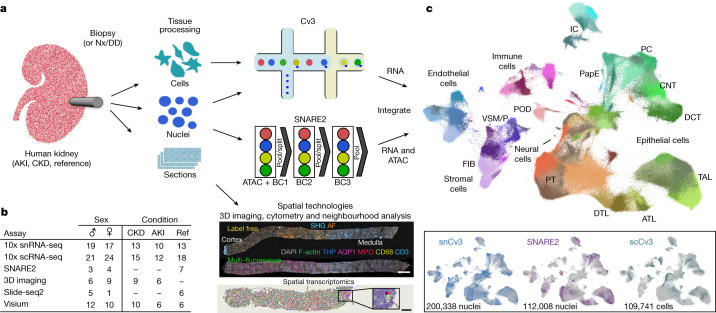
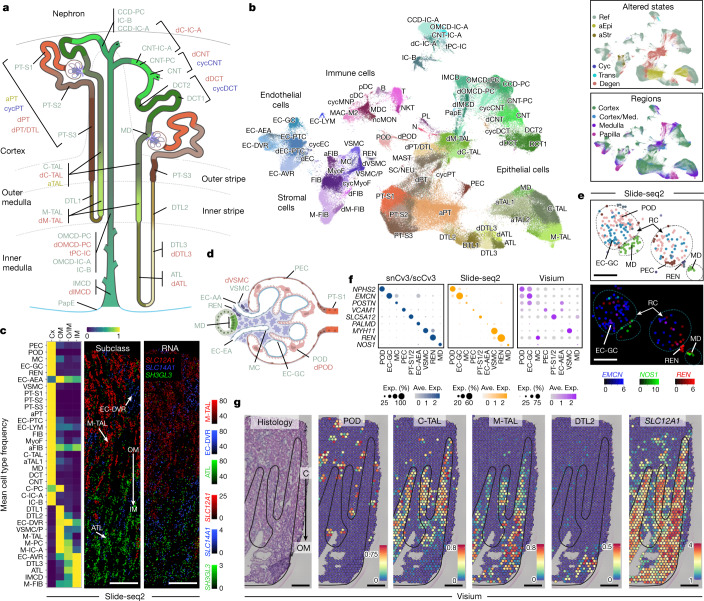
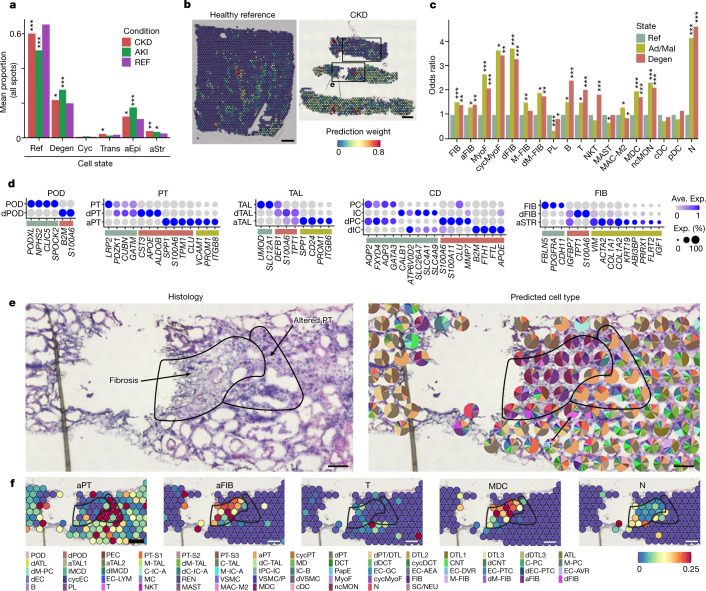
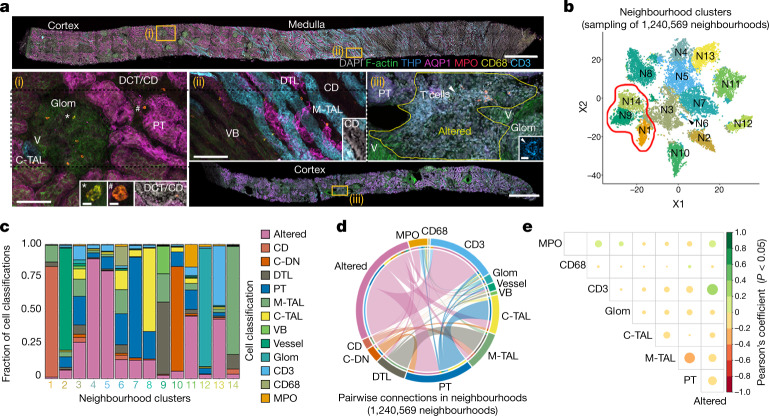
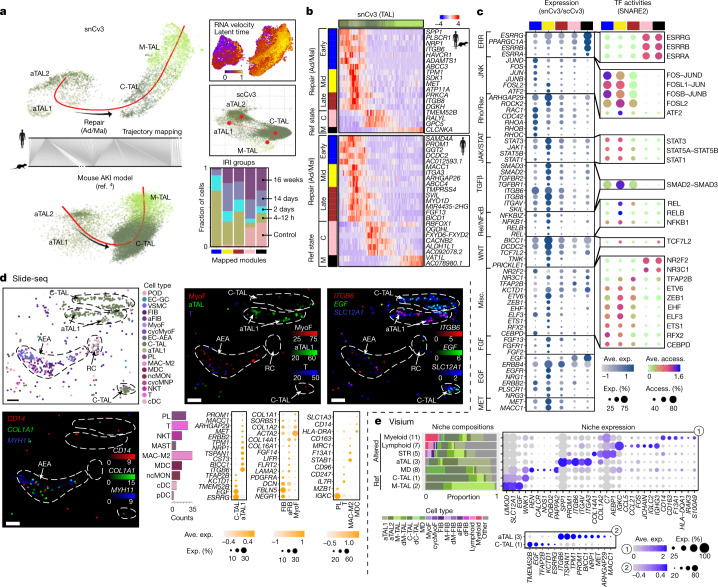
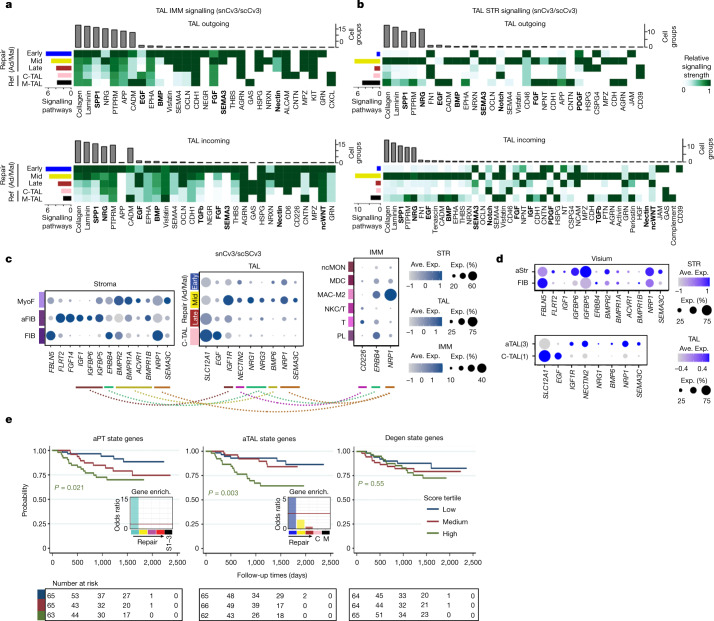
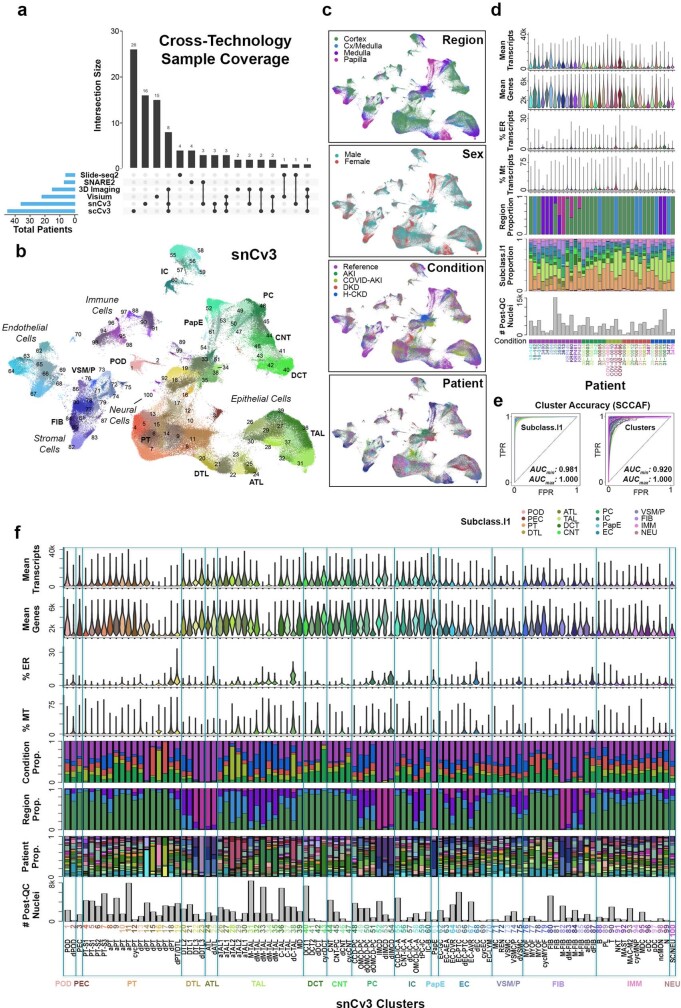
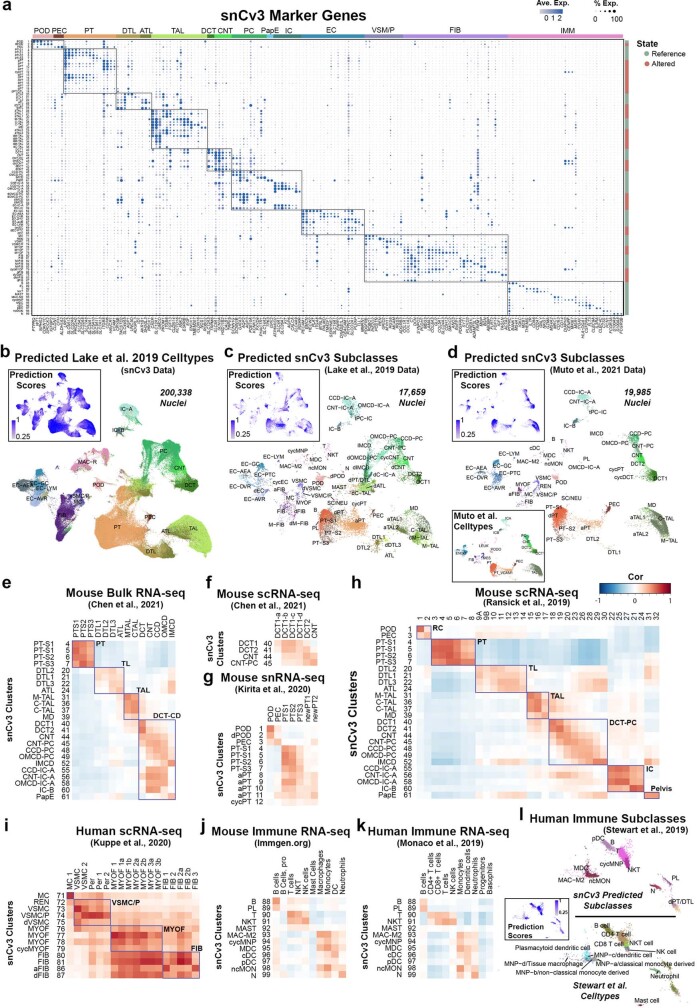
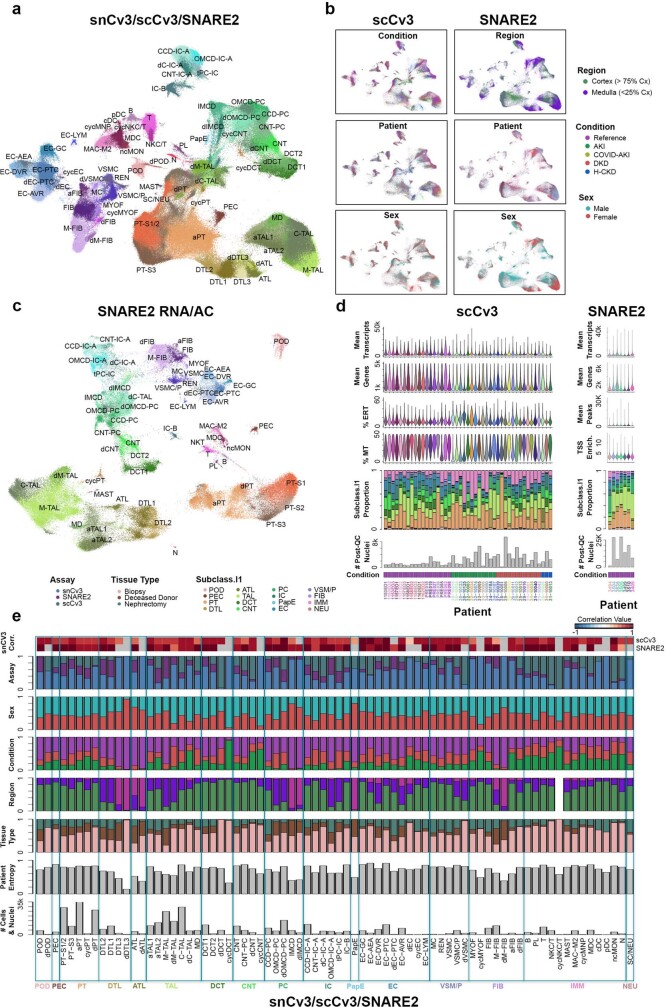
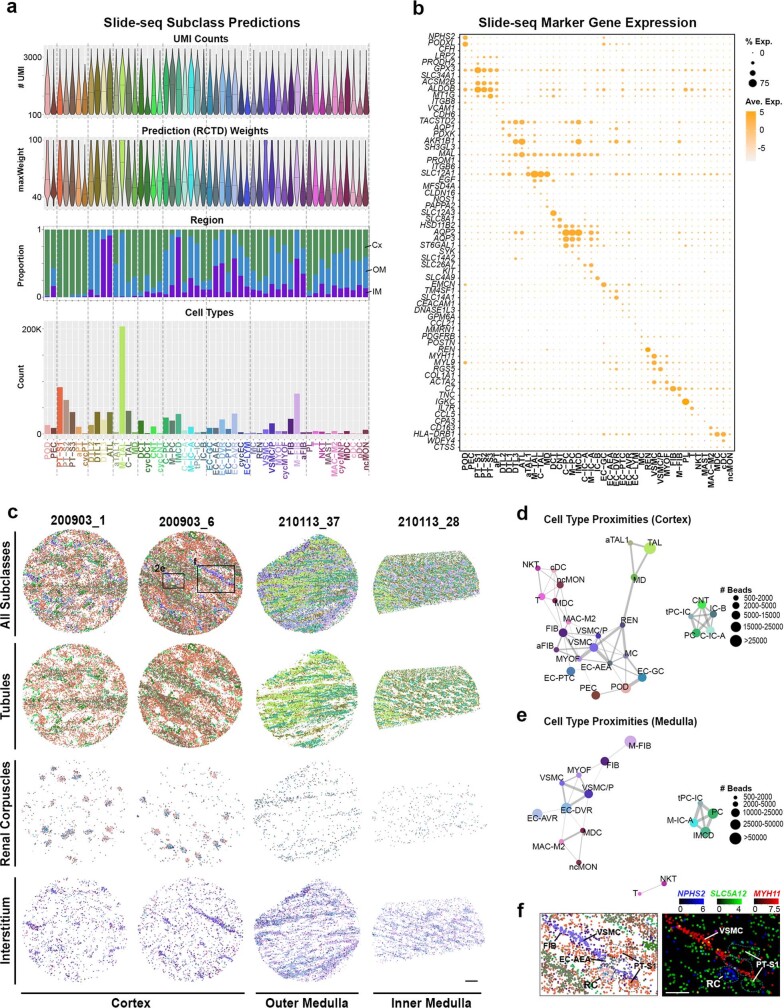
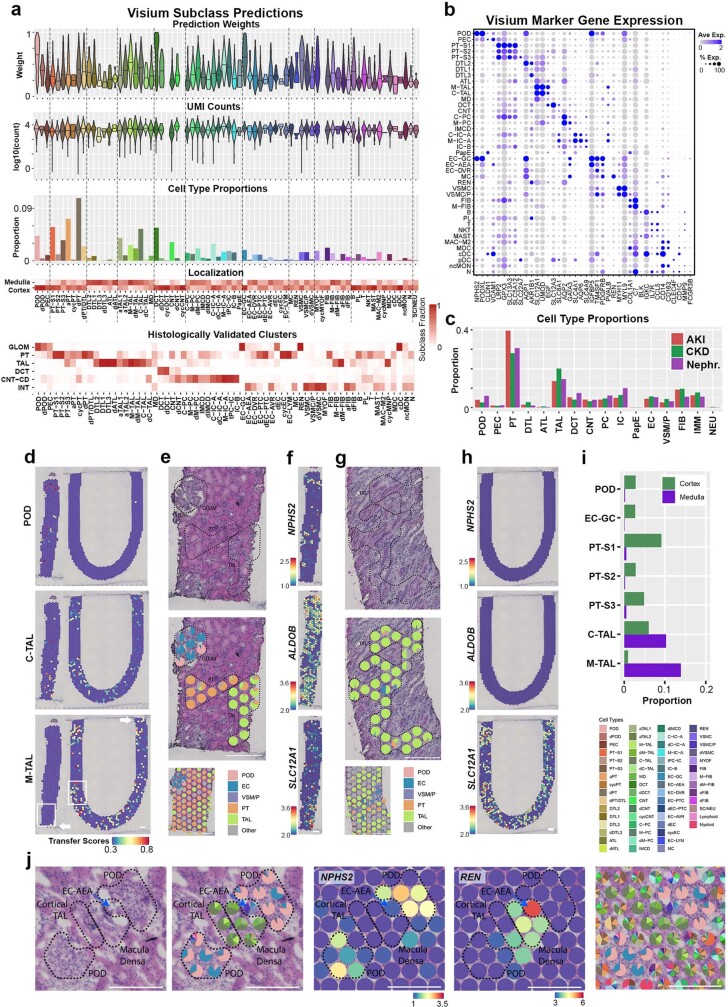
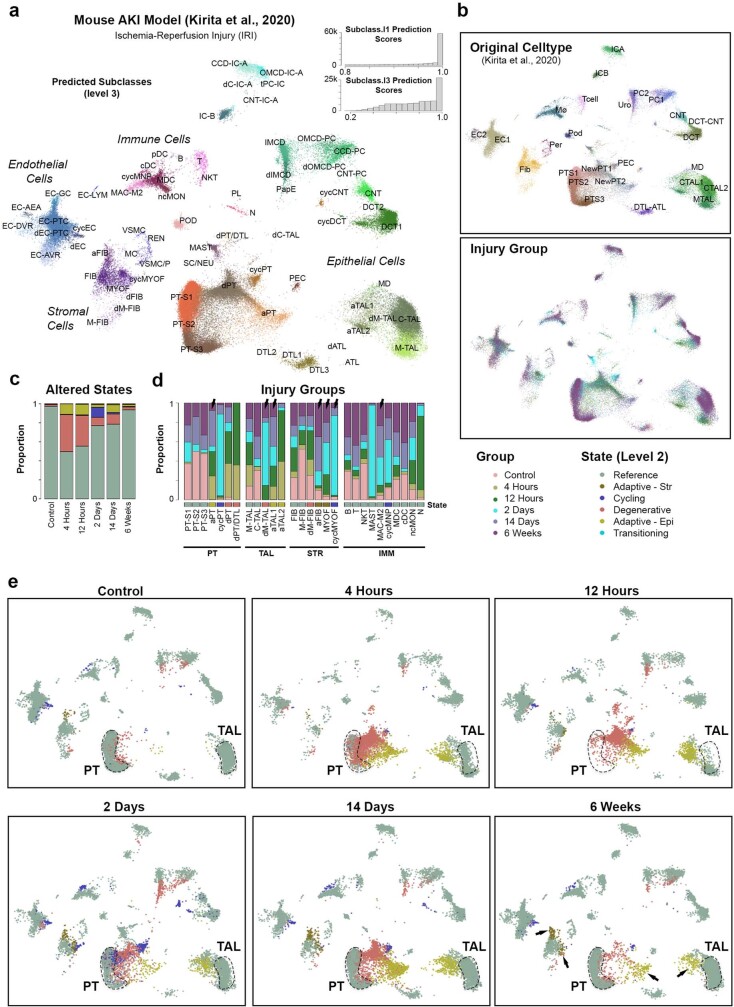
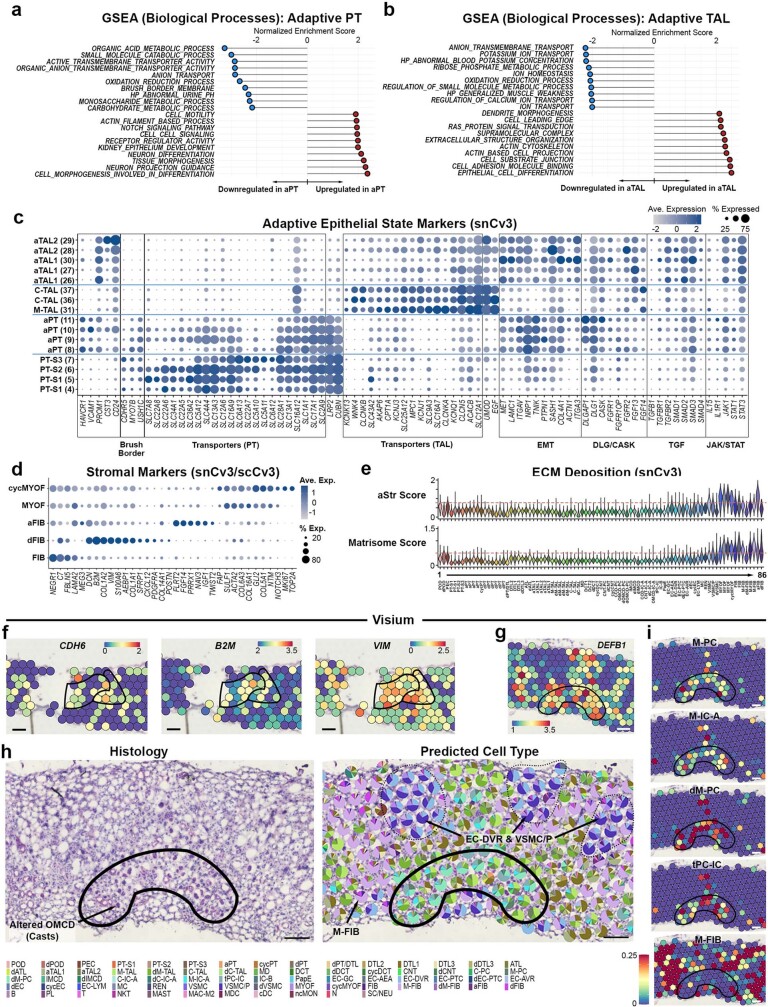
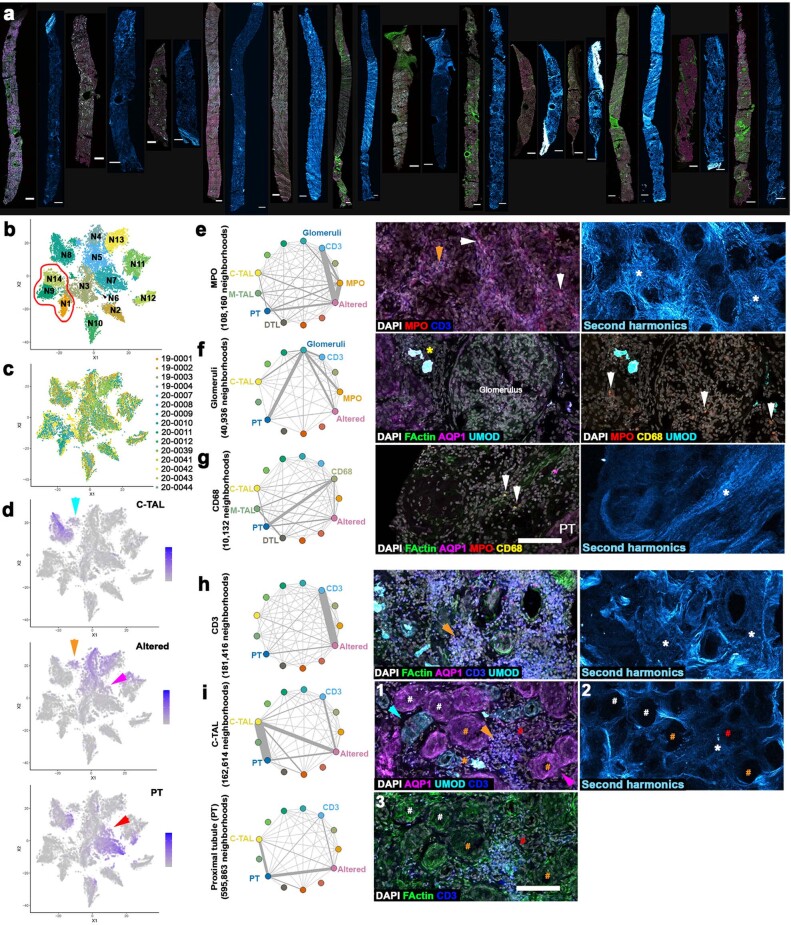
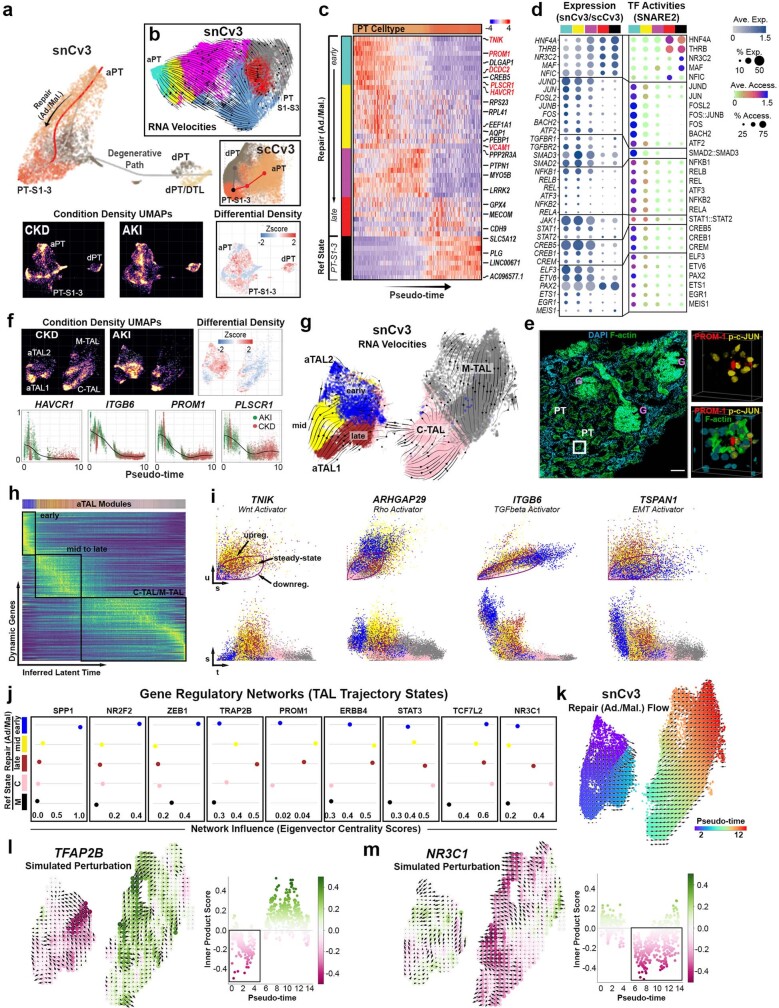
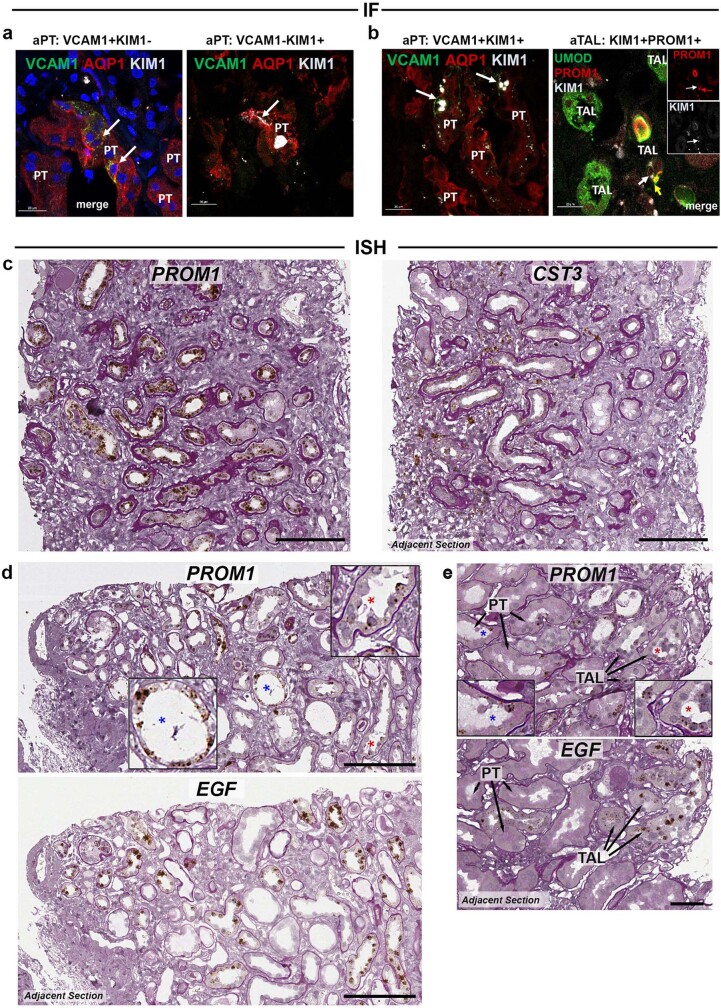
Understanding kidney disease relies on defining the complexity of cell types and states, their associated molecular profiles and interactions within tissue neighbourhoods1. Here we applied multiple single-cell and single-nucleus assays (>400,000 nuclei or cells) and spatial imaging technologies to a broad spectrum of healthy reference kidneys (45 donors) and diseased kidneys (48 patients). This has provided a high-resolution cellular atlas of 51 main cell types, which include rare and previously undescribed cell populations. The multi-omic approach provides detailed transcriptomic profiles, regulatory factors and spatial localizations spanning the entire kidney. We also define 28 cellular states across nephron segments and interstitium that were altered in kidney injury, encompassing cycling, adaptive (successful or maladaptive repair), transitioning and degenerative states. Molecular signatures permitted the localization of these states within injury neighbourhoods using spatial transcriptomics, while large-scale 3D imaging analysis (around 1.2 million neighbourhoods) provided corresponding linkages to active immune responses. These analyses defined biological pathways that are relevant to injury time-course and niches, including signatures underlying epithelial repair that predicted maladaptive states associated with a decline in kidney function. This integrated multimodal spatial cell atlas of healthy and diseased human kidneys represents a comprehensive benchmark of cellular states, neighbourhoods, outcome-associated signatures and publicly available interactive visualizations.
© 2023. The Author(s).
Conflict of interest statement
P.V.K. serves on the scientific advisory board to Celsius Therapeutics and Biomage. A.V. is a consultant for Astute and NxStage. C.R.P. is a member of the advisory board of and owns equity in RenalytixAI, and serves as a consultant for Genfit and Novartis. M.K. has grants from JDRF, Astra-Zeneca, NovoNordisc, Eli Lilly, Gilead, Goldfinch Bio, Janssen, Boehringer-Ingelheim, Moderna, European Union Innovative Medicine Initiative, Chan Zuckerberg Initiative, Certa, Chinook, amfAR, Angion Pharmaceuticals, RenalytixAI, Travere Therapeutics, Regeneron, IONIS Pharmaceuticals, Astellas, Poxel and a patent (PCT/EP2014/073413; ‘Biomarkers and methods for progression prediction for chronic kidney disease’) licensed. F.C. and E.Z.M. are paid consultants for Atlas Bio. F.P.W. receives research support from Astrazeneca, Boeringher-Ingelheim, Vifor Pharma and Whoop. P.M.P. is a consultant for Janssen. S.R. has research funding from AstraZeneca and Bayer Healthcare. S.S.W. is a consultant for GSK, GEHC, JNJ, Strataca, Roth Capital Partners, Venbio, and an expert witness on litigation for Davita and Pfizer. J.R.S. consults for Maze and Goldfinch and receives royalties from Sanfi Genzyme. K.Z. is a co-founder, equity holder and serves on the scientific advisory board of Singlera Genomics. A.S.N. is on the external advisory board for CareDX. L.H.M. is a consultant for Reata Pharmaceuticals, Travere Therapeutics and Calliditas. S.J. is a paid Blue SKy mentor for Meharry Medical College, Nashville and receives royalties from Elsevier. J.L.M. is an employee and shareholder of Solid Biosciences. The other authors declare no competing interests.
Figures
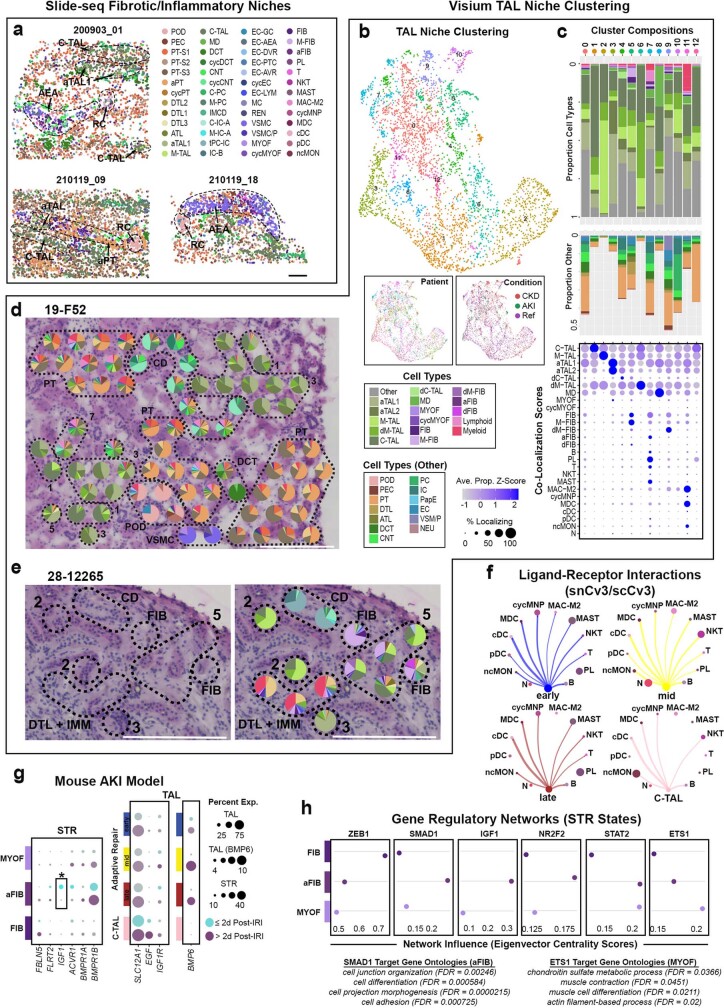

References
Publication types
MeSH terms
Grants and funding
- U01 DK114923/DK/NIDDK NIH HHS/United States
- UH3 DK114907/DK/NIDDK NIH HHS/United States
- K08 DK107864/DK/NIDDK NIH HHS/United States
- UH3 DK114908/DK/NIDDK NIH HHS/United States
- U2C DK114886/DK/NIDDK NIH HHS/United States
- U54 DK083912/DK/NIDDK NIH HHS/United States
- UH3 DK114861/DK/NIDDK NIH HHS/United States
- U54 HL145608/HL/NHLBI NIH HHS/United States
- UH3 DK114866/DK/NIDDK NIH HHS/United States
- UH3 DK114926/DK/NIDDK NIH HHS/United States
- UL1 TR002319/TR/NCATS NIH HHS/United States
- U54 DK134301/DK/NIDDK NIH HHS/United States
- UL1 TR002345/TR/NCATS NIH HHS/United States
- U01 MH114828/MH/NIMH NIH HHS/United States
- UH3 CA246632/CA/NCI NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- UH3 DK114923/DK/NIDDK NIH HHS/United States
- K23 DK125529/DK/NIDDK NIH HHS/United States
- U01 DK133090/DK/NIDDK NIH HHS/United States
- UH3 DK114915/DK/NIDDK NIH HHS/United States
- U01 DK114933/DK/NIDDK NIH HHS/United States
- P30 DK036836/DK/NIDDK NIH HHS/United States
- S10 OD026929/OD/NIH HHS/United States
- P30 DK079312/DK/NIDDK NIH HHS/United States
- P30 CA091842/CA/NCI NIH HHS/United States
- UH3 DK114870/DK/NIDDK NIH HHS/United States
- U01 DK114907/DK/NIDDK NIH HHS/United States
- UG3 DK114861/DK/NIDDK NIH HHS/United States
- U54 DK137328/DK/NIDDK NIH HHS/United States
- P30 DK081943/DK/NIDDK NIH HHS/United States
- R01 DK111651/DK/NIDDK NIH HHS/United States
- UH3 DK114933/DK/NIDDK NIH HHS/United States
- P01 DK056788/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
