

KRAS(G12D) drives lepidic adenocarcinoma through stem-cell reprogramming
- PMID: 37468622
- PMCID: PMC10423036
- DOI: 10.1038/s41586-023-06324-w
KRAS(G12D) drives lepidic adenocarcinoma through stem-cell reprogramming
Abstract
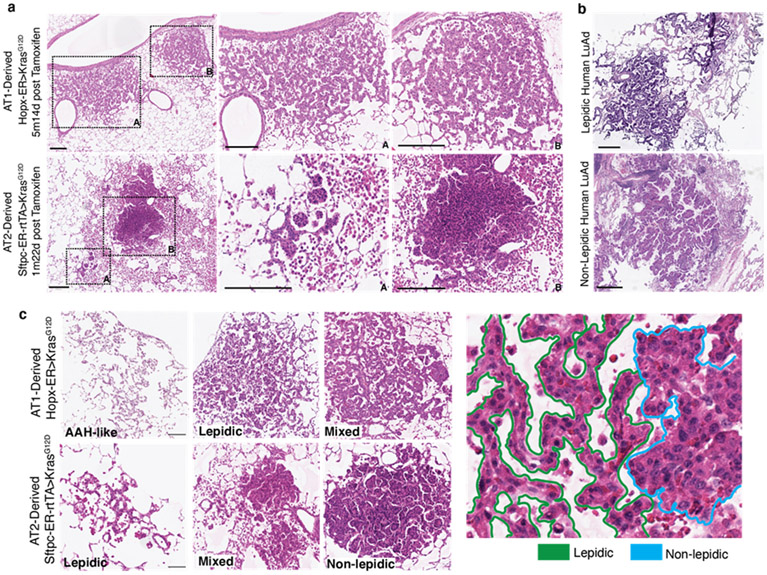
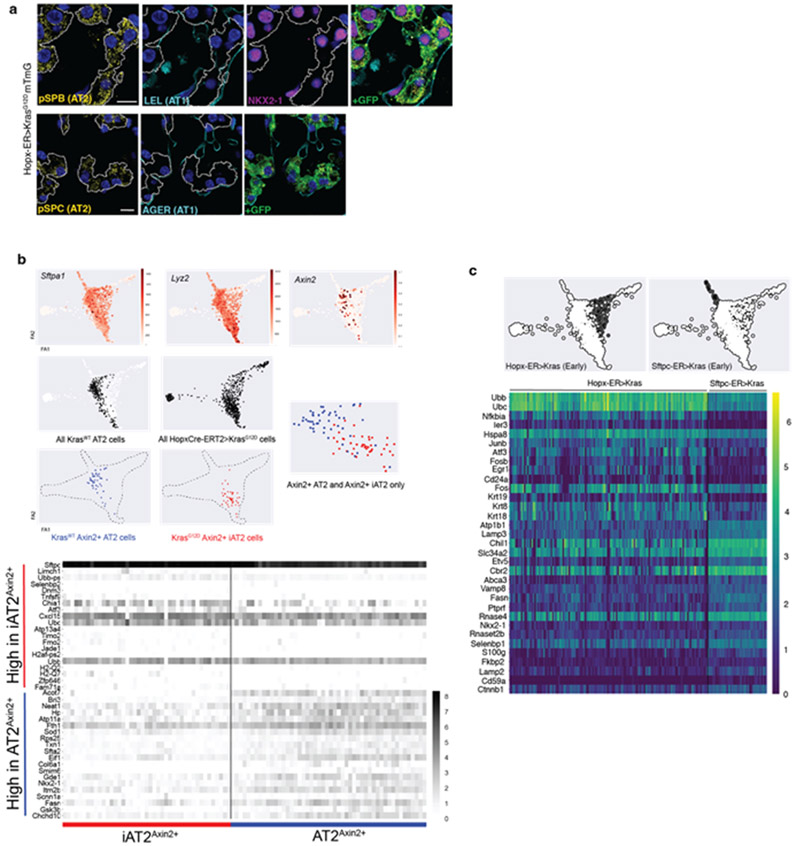
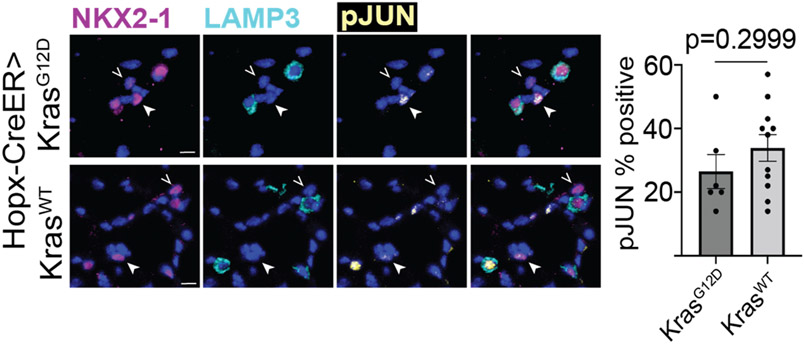
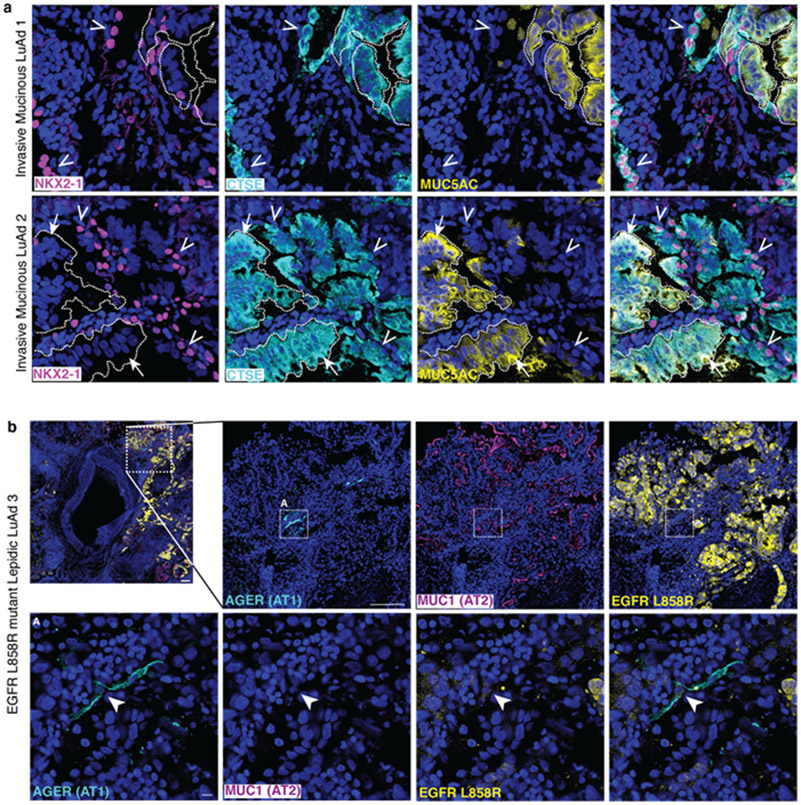
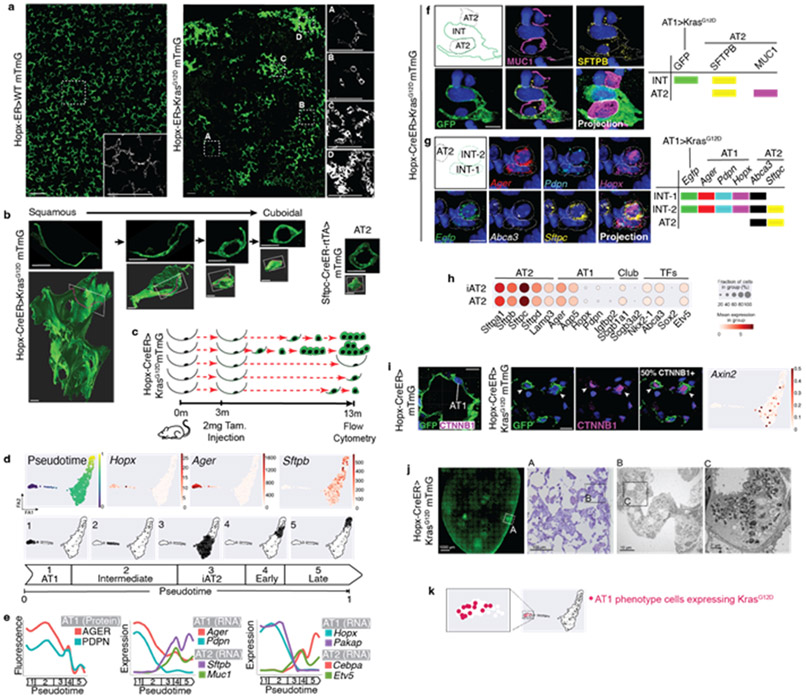
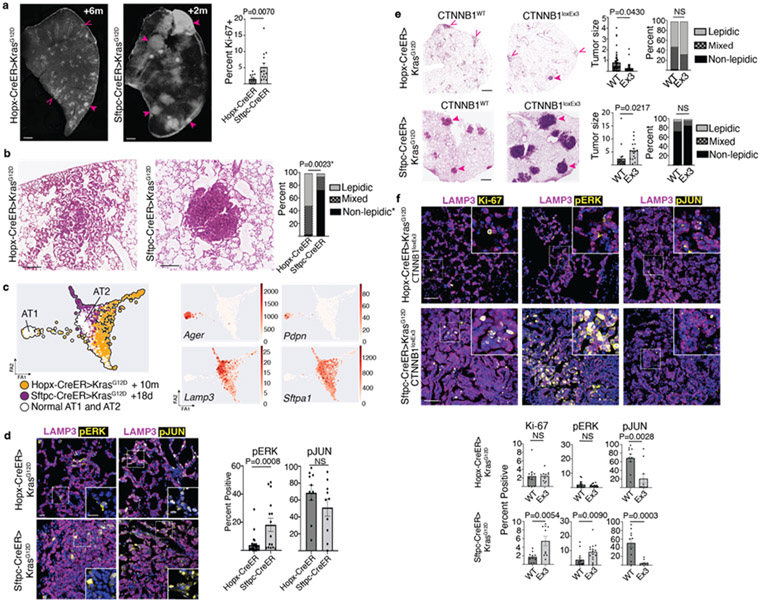
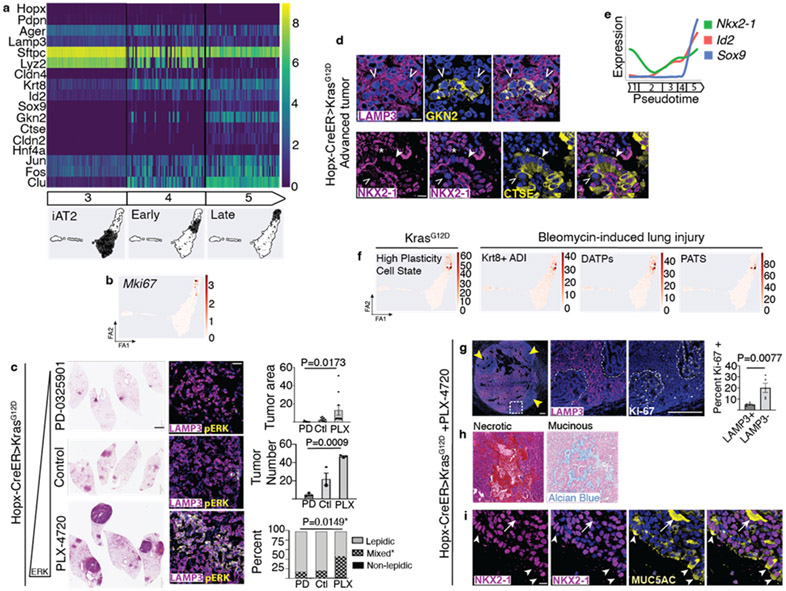
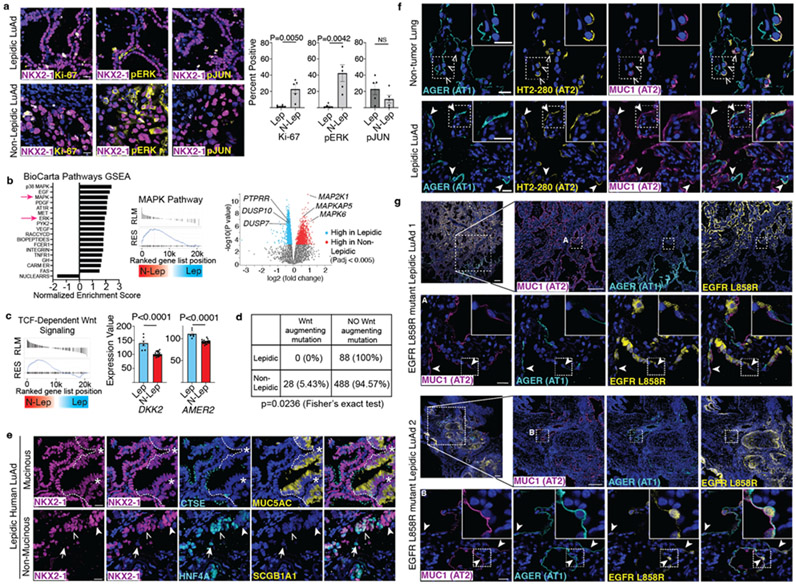
Many cancers originate from stem or progenitor cells hijacked by somatic mutations that drive replication, exemplified by adenomatous transformation of pulmonary alveolar epithelial type II (AT2) cells1. Here we demonstrate a different scenario: expression of KRAS(G12D) in differentiated AT1 cells reprograms them slowly and asynchronously back into AT2 stem cells that go on to generate indolent tumours. Like human lepidic adenocarcinoma, the tumour cells slowly spread along alveolar walls in a non-destructive manner and have low ERK activity. We find that AT1 and AT2 cells act as distinct cells of origin and manifest divergent responses to concomitant WNT activation and KRAS(G12D) induction, which accelerates AT2-derived but inhibits AT1-derived adenoma proliferation. Augmentation of ERK activity in KRAS(G12D)-induced AT1 cells increases transformation efficiency, proliferation and progression from lepidic to mixed tumour histology. Overall, we have identified a new cell of origin for lung adenocarcinoma, the AT1 cell, which recapitulates features of human lepidic cancer. In so doing, we also uncover a capacity for oncogenic KRAS to reprogram a differentiated and quiescent cell back into its parent stem cell en route to adenomatous transformation. Our work further reveals that irrespective of a given cancer's current molecular profile and driver oncogene, the cell of origin exerts a pervasive and perduring influence on its subsequent behaviour.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
Comment in
-
Mutation-Driven Alveolar Cell Reprogramming Dictates Lung Cancer Progression.Cancer Discov. 2023 Sep 6;13(9):OF7. doi: 10.1158/2159-8290.CD-RW2023-117. Cancer Discov. 2023. PMID: 37504795
References
-
- Desai TJ Developmental Insights into Lung Cancer. Annual review of cancer biology 5, 351–369 (2021). 10.1146/annurev-cancerbio-070820-032858 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
