

Extreme dynamics in a biomolecular condensate
- PMID: 37468629
- PMCID: PMC11508043
- DOI: 10.1038/s41586-023-06329-5
Extreme dynamics in a biomolecular condensate
Abstract
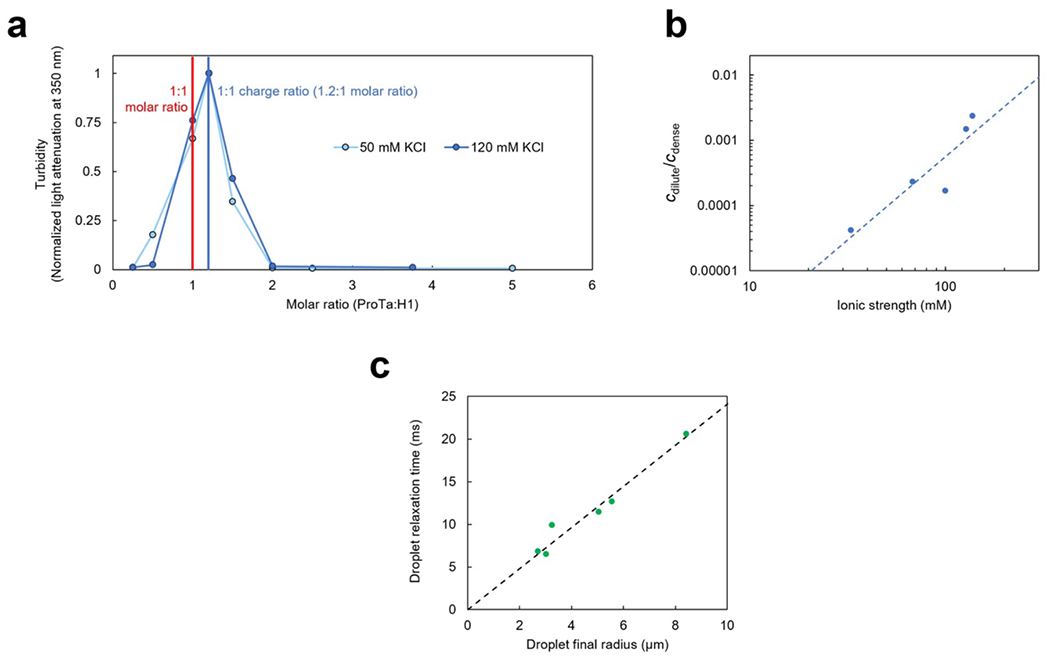
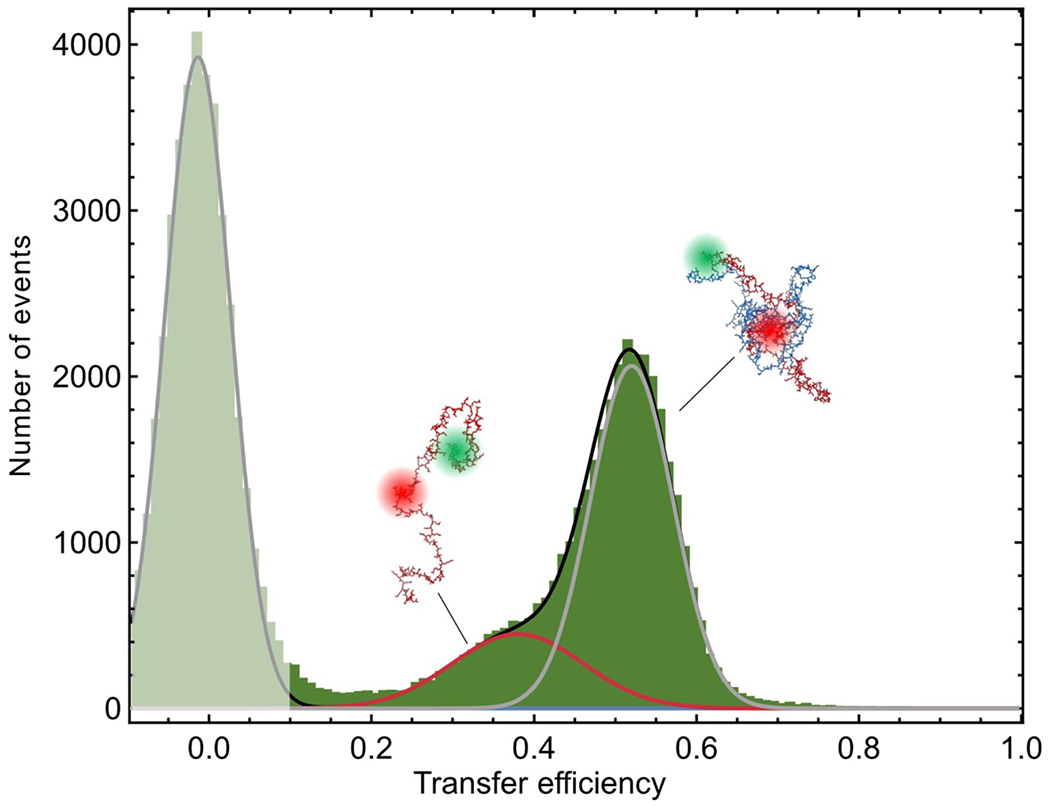
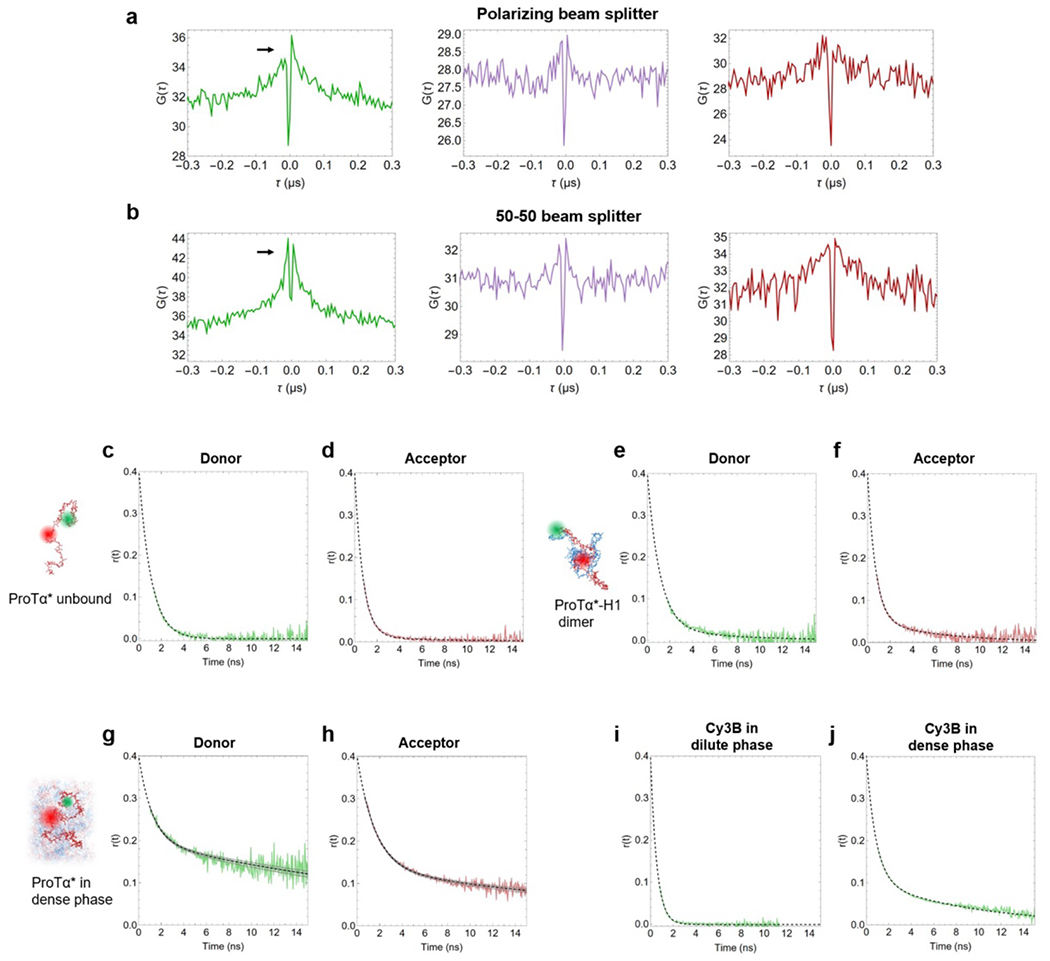
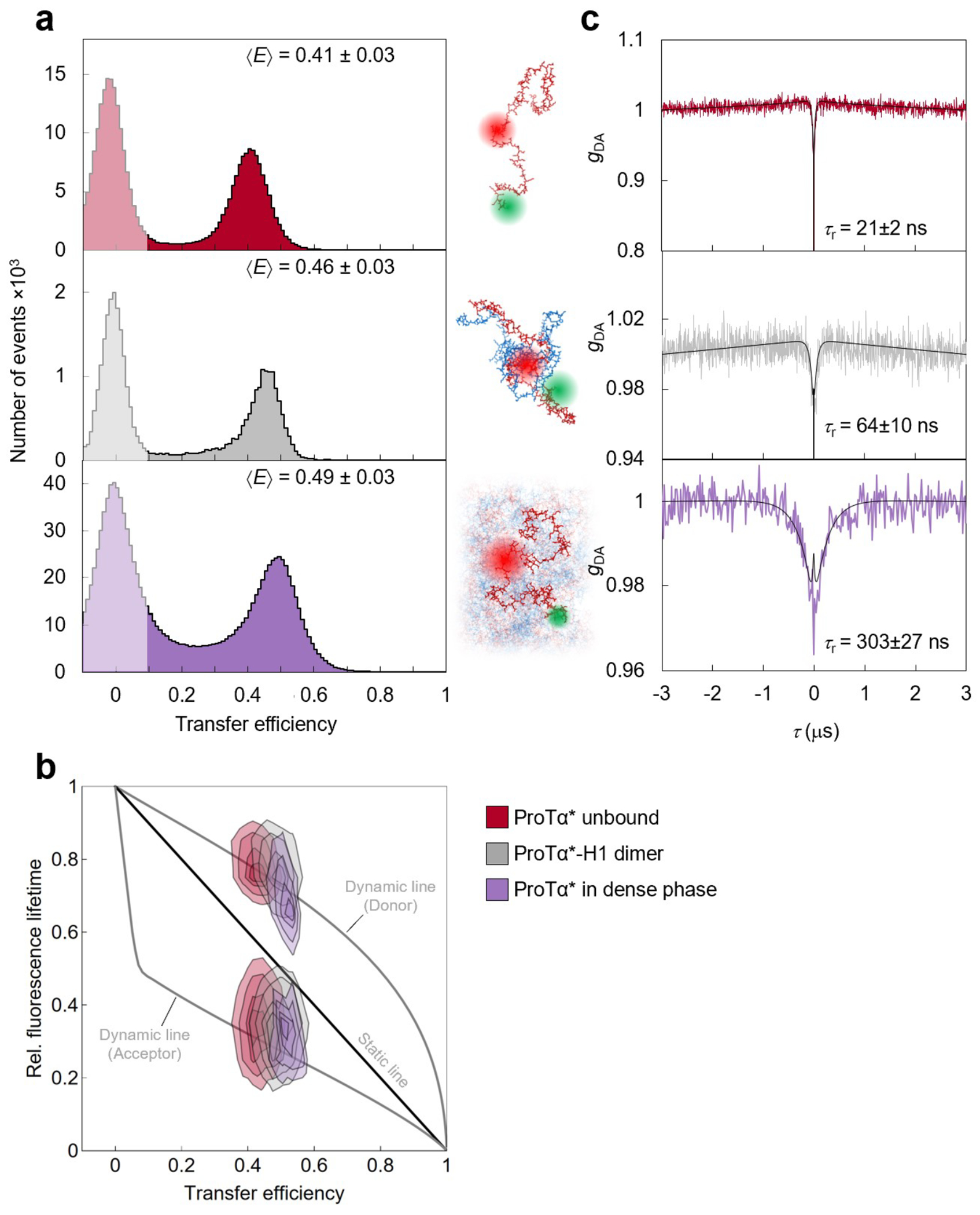
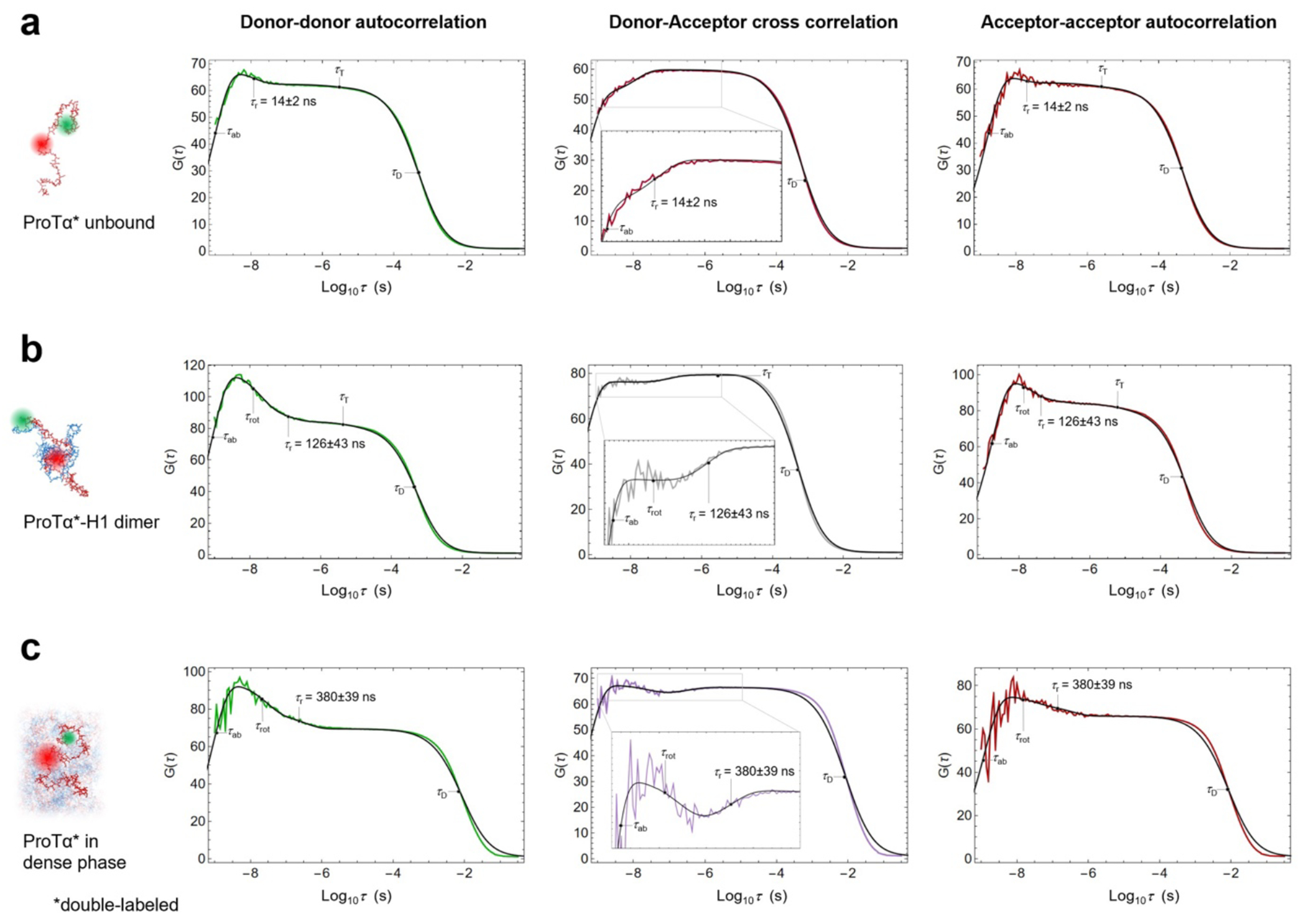
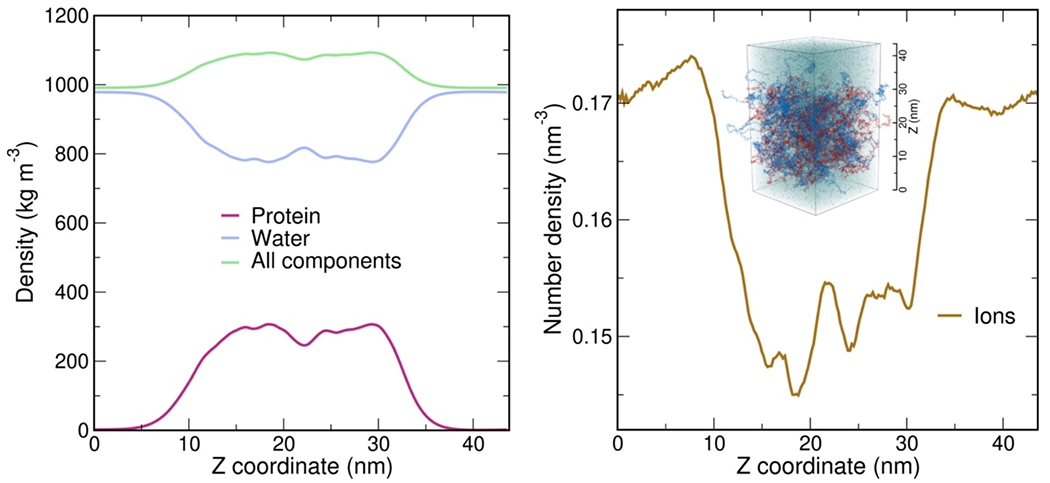
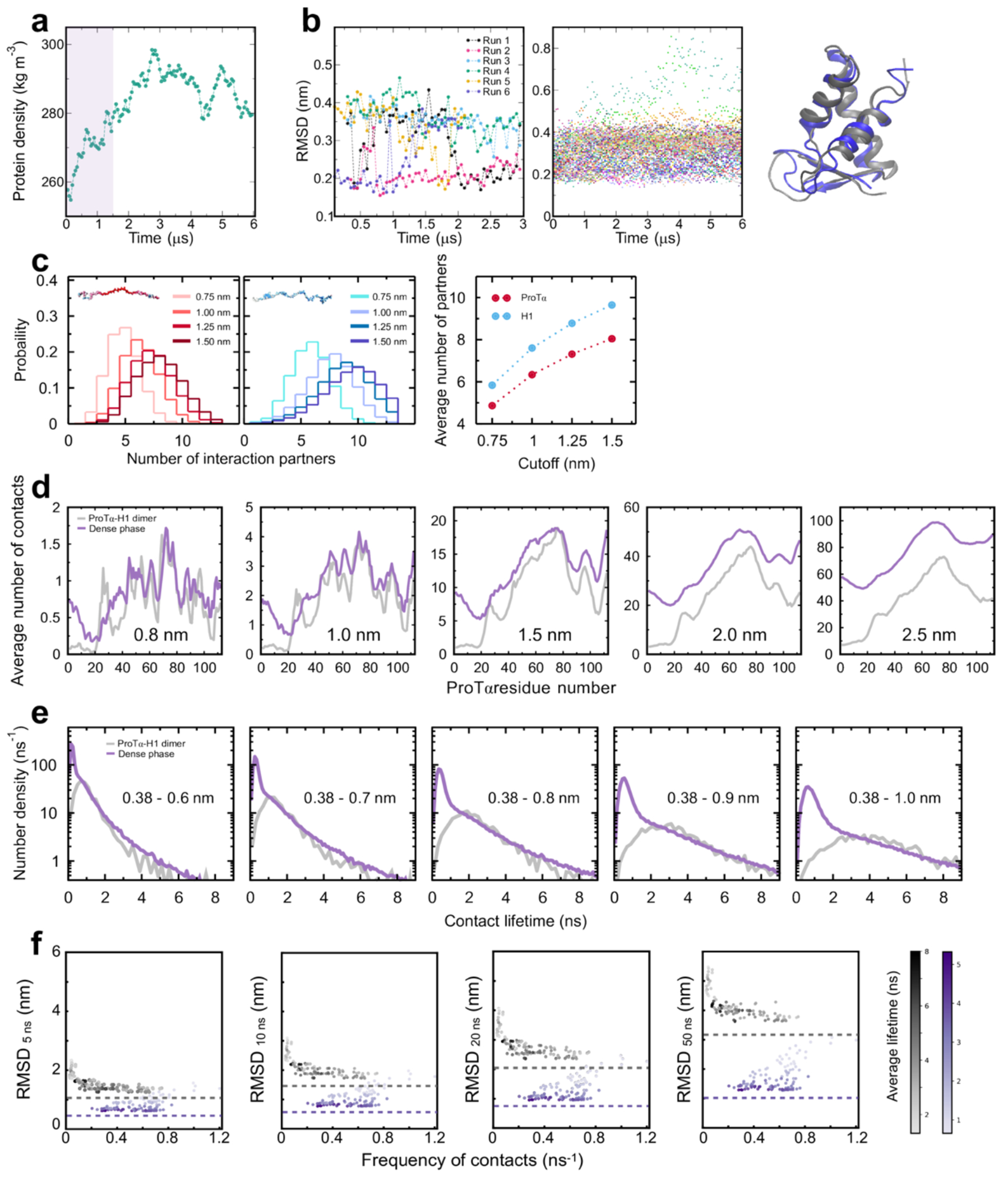
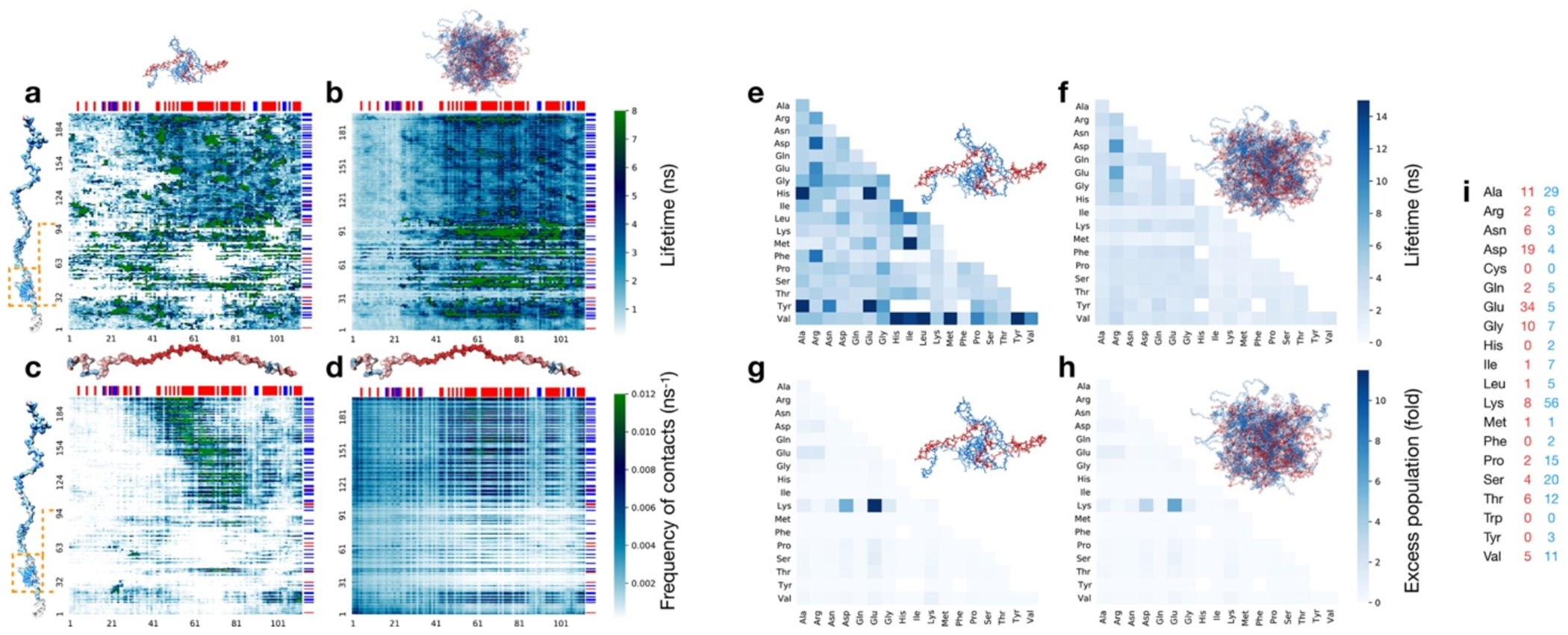
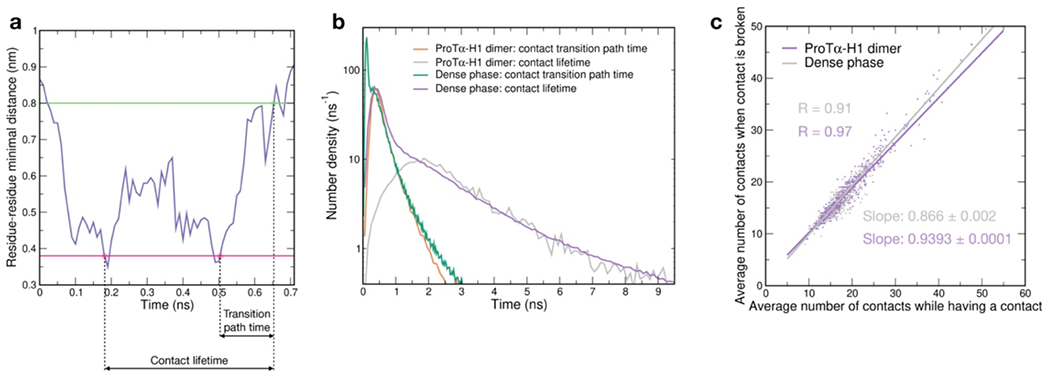
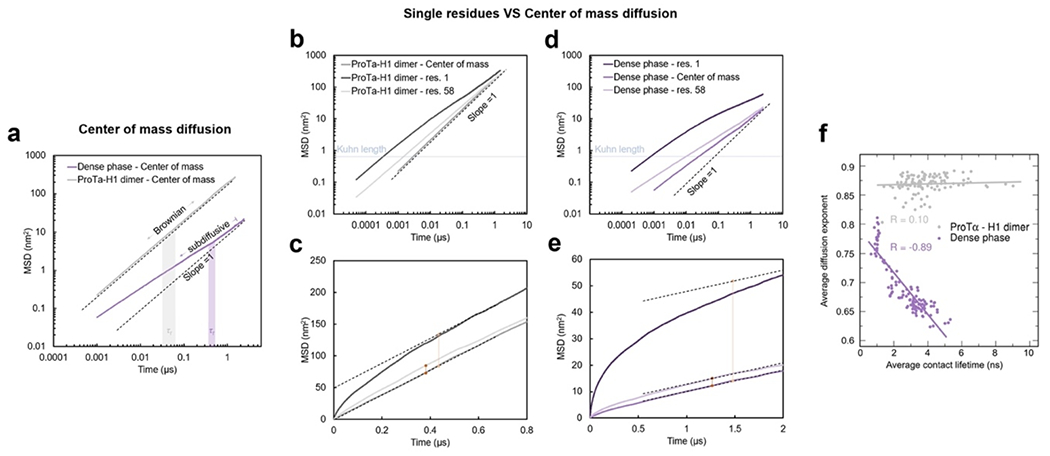
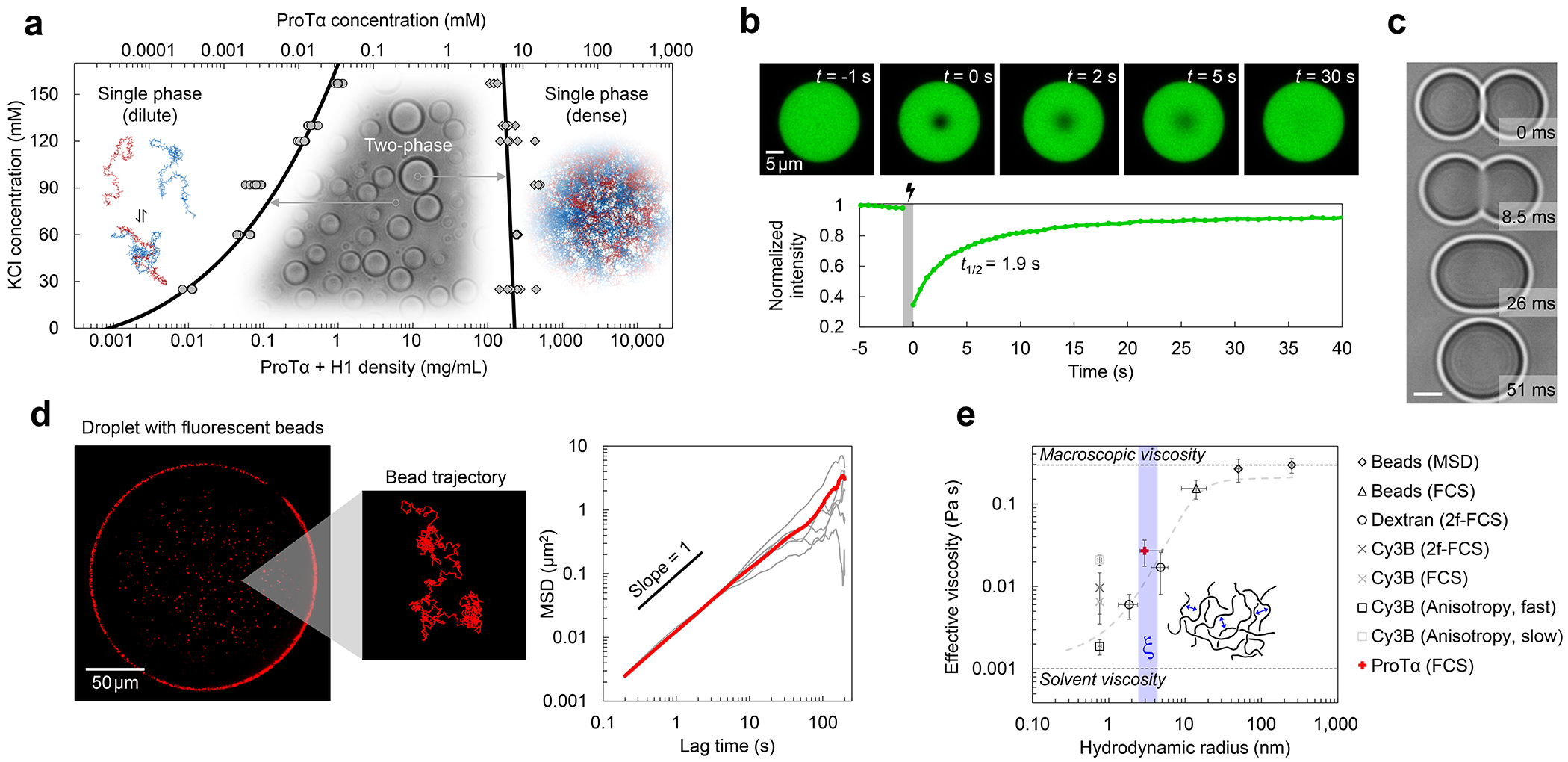
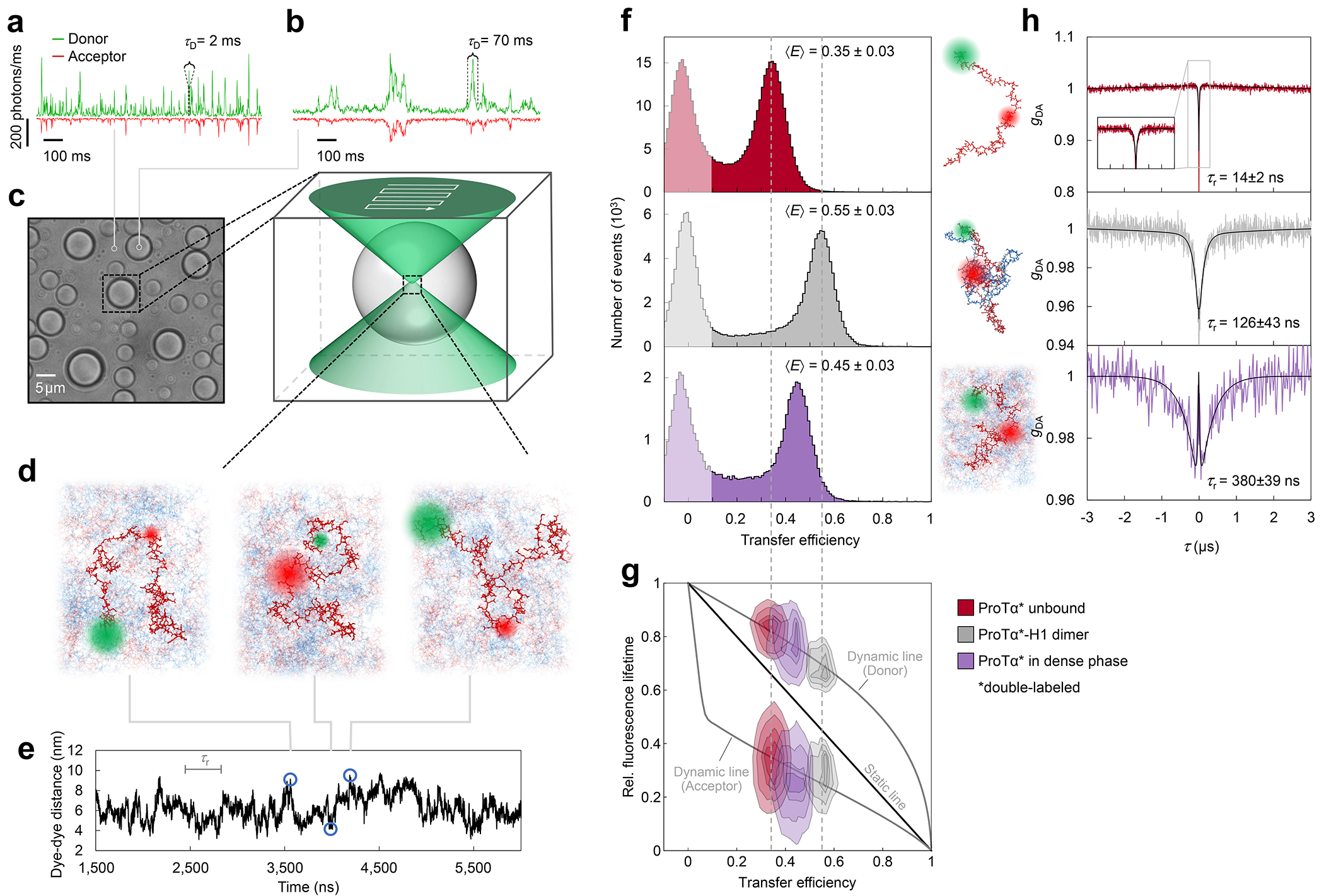
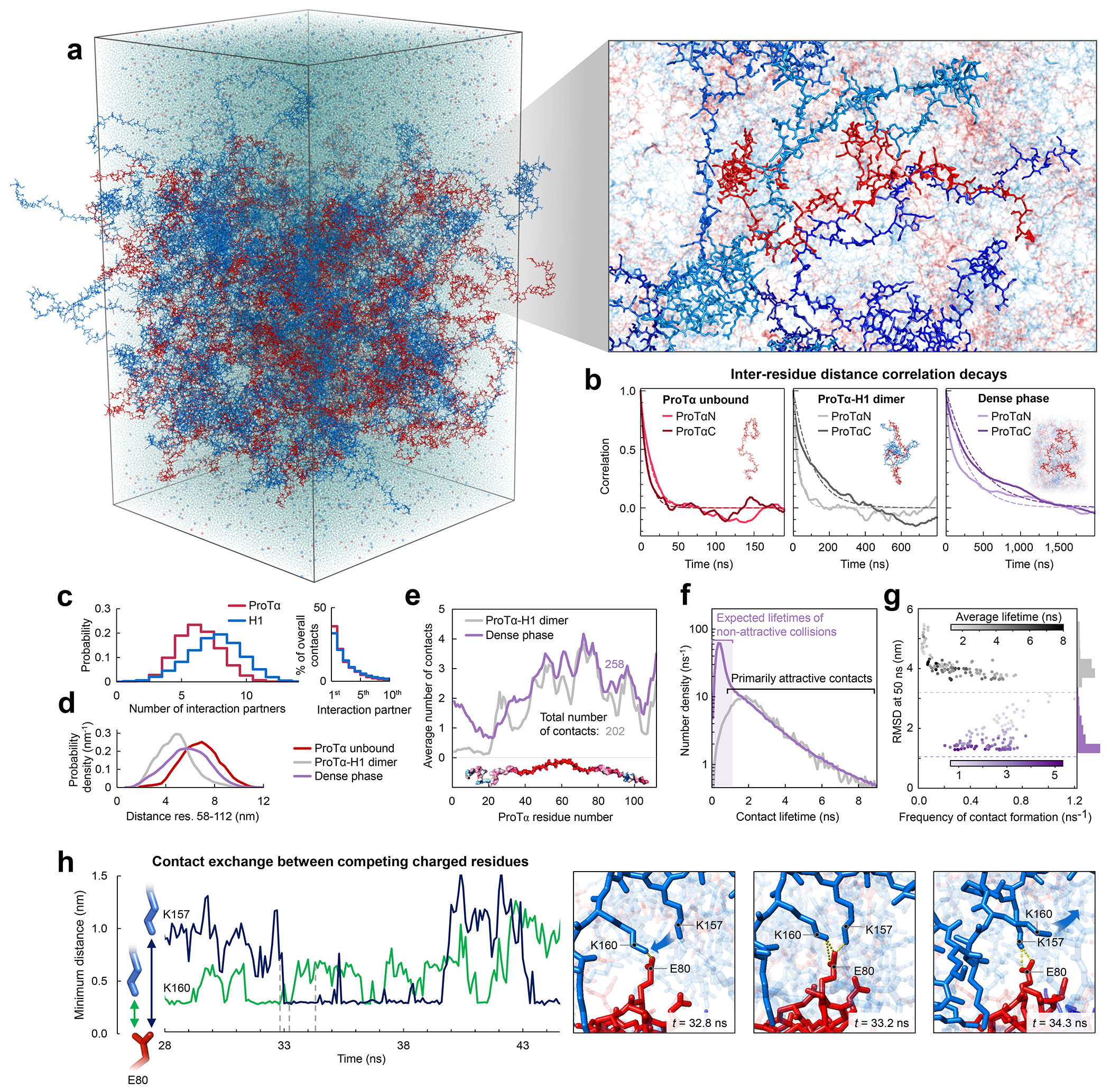
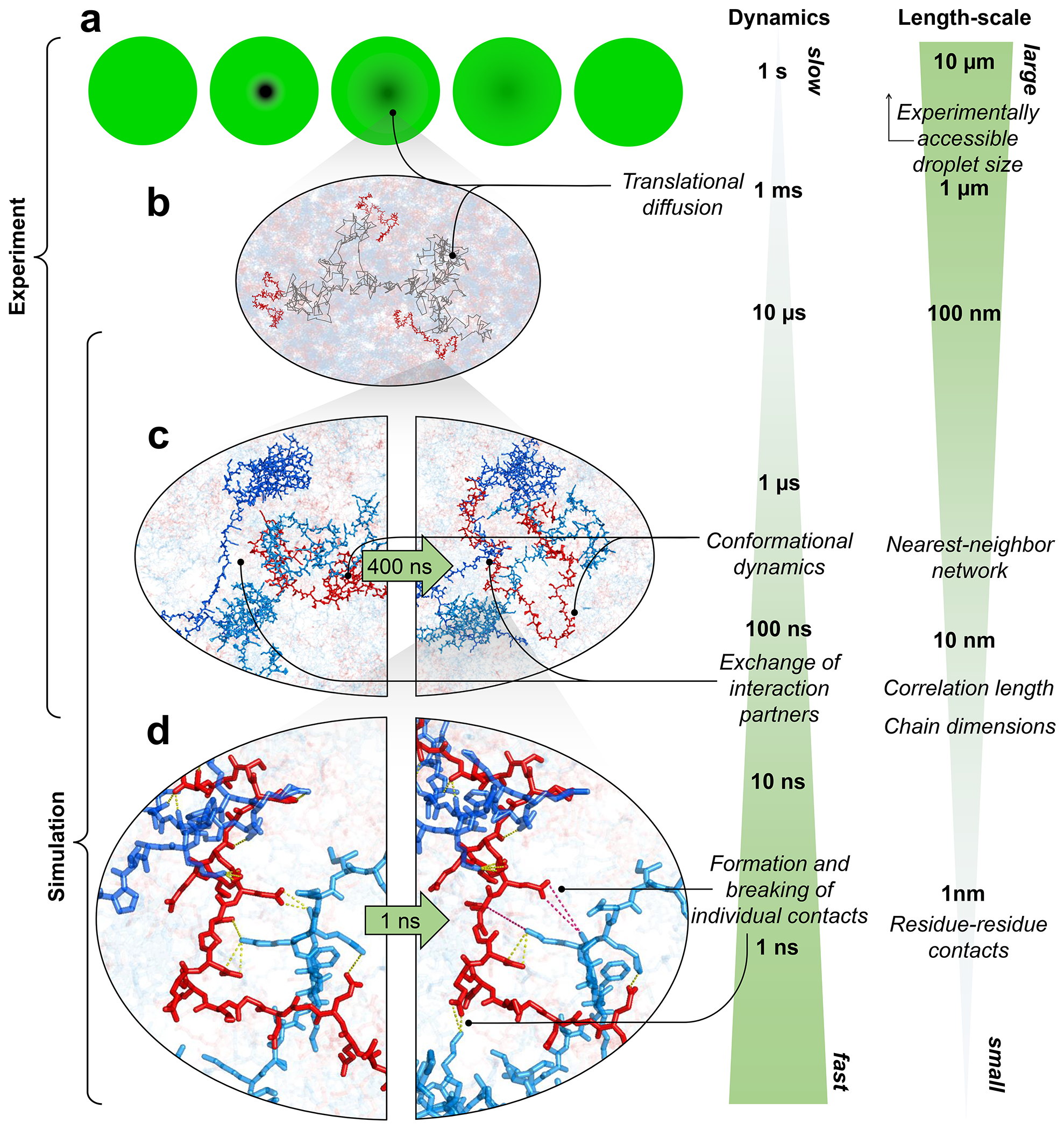
Proteins and nucleic acids can phase-separate in the cell to form concentrated biomolecular condensates1-4. The functions of condensates span many length scales: they modulate interactions and chemical reactions at the molecular scale5, organize biochemical processes at the mesoscale6 and compartmentalize cells4. Understanding the underlying mechanisms of these processes will require detailed knowledge of the rich dynamics across these scales7. The mesoscopic dynamics of biomolecular condensates have been extensively characterized8, but their behaviour at the molecular scale has remained more elusive. Here, as an example of biomolecular phase separation, we study complex coacervates of two highly and oppositely charged disordered human proteins9. Their dense phase is 1,000 times more concentrated than the dilute phase, and the resulting percolated interaction network10 leads to a bulk viscosity 300 times greater than that of water. However, single-molecule spectroscopy optimized for measurements within individual droplets reveals that at the molecular scale, the disordered proteins remain exceedingly dynamic, with their chain configurations interconverting on submicrosecond timescales. Massive all-atom molecular dynamics simulations reproduce the experimental observations and explain this apparent discrepancy: the underlying interactions between individual charged side chains are short-lived and exchange on a pico- to nanosecond timescale. Our results indicate that, despite the high macroscopic viscosity of phase-separated systems, local biomolecular rearrangements required for efficient reactions at the molecular scale can remain rapid.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Comment in
-
Dynamics of protein droplets revealed by bridging multiple scales.Nature. 2023 Jul;619(7971):700-701. doi: 10.1038/d41586-023-02215-2. Nature. 2023. PMID: 37468813 No abstract available.
References
-
- Abascal JLF, and Vega C. 2005. ‘A general purpose model for the condensed phases of water: TIP4P/2005’, J. Chem. Phys, 123: 234505. - PubMed
-
- Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, and Lindahl E. 2015. ‘GROMACS: High performance molecular simulations through multi-levelvparallelism from laptops to supercomputers’, SoftwareX, 1-2: 19–25.
-
- Alberts Bruce. 2022. Molecular biology of the cell (W. W. Norton & Company: New York: ).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
