

Phenotype Presentation and Molecular Diagnostic Yield in Non-5q Spinal Muscular Atrophy
- PMID: 37470033
- PMCID: PMC10352921
- DOI: 10.1212/NXG.0000000000200087
Phenotype Presentation and Molecular Diagnostic Yield in Non-5q Spinal Muscular Atrophy
Erratum in
-
Erratum: Phenotype Presentation and Molecular Diagnostic Yield in Non-5q Spinal Muscular Atrophy.Neurol Genet. 2024 Jan 10;10(1):e200123. doi: 10.1212/NXG.0000000000200123. eCollection 2024 Feb. Neurol Genet. 2024. PMID: 38213751 Free PMC article.
-
Erratum: Phenotype Presentation and Molecular Diagnostic Yield in Non-5q Spinal Muscular Atrophy.Neurol Genet. 2024 Apr 25;10(3):e200139. doi: 10.1212/NXG.0000000000200139. eCollection 2024 Jun. Neurol Genet. 2024. PMID: 38685977 Free PMC article.
Abstract
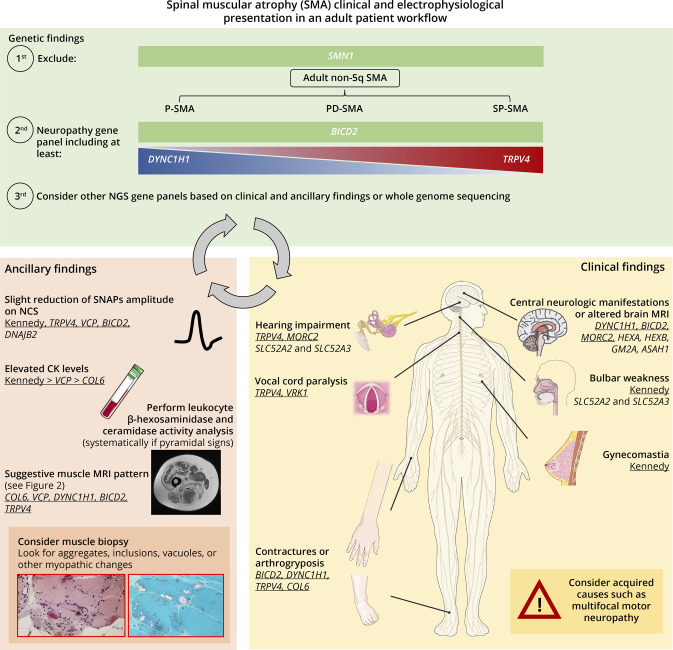
Background and objectives: Spinal muscular atrophy (SMA) is mainly caused by homozygous SMN1 gene deletions on 5q13. Non-5q SMA patients' series are lacking, and the diagnostic yield of next-generation sequencing (NGS) is largely unknown. The aim of this study was to describe the clinical and genetic landscape of non-5q SMA and evaluate the performance of neuropathy gene panels in these disorders.
Methods: Description of patients with non-5q SMA followed in the different neuromuscular reference centers in France as well as in London, United Kingdom. Patients without a genetic diagnosis had undergone at least a neuropathy or large neuromuscular gene panel.
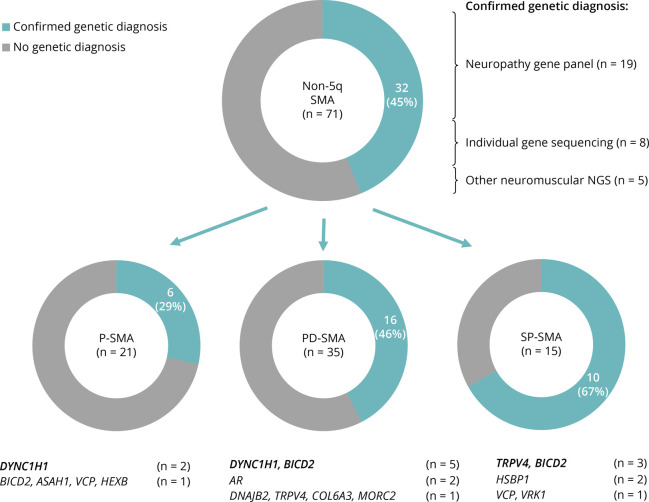
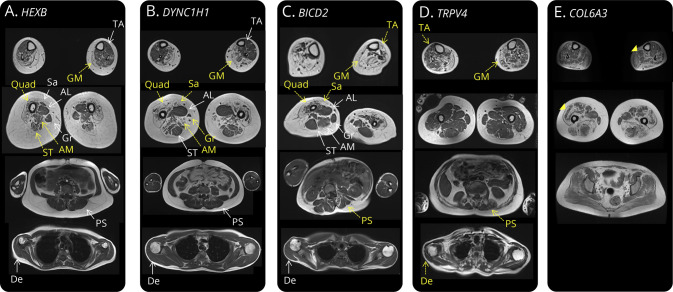
Results: Seventy-one patients from 65 different families were included, mostly sporadic cases (60.6%). At presentation, 21 patients (29.6%) showed exclusive proximal weakness (P-SMA), 35 (49.3%) showed associated distal weakness (PD-SMA), and 15 (21.1%) a scapuloperoneal phenotype (SP-SMA). Thirty-two patients (45.1%) had a genetic diagnosis: BICD2 (n = 9), DYNC1H1 (n = 7), TRPV4 (n = 4), VCP, HSBP1, AR (n = 2), VRK1, DNAJB2, MORC2, ASAH1, HEXB, and unexpectedly, COL6A3 (n = 1). The genetic diagnostic yield was lowest in P-SMA (6/21, 28.6%) compared with PD-SMA (16/35, 45.7%) and SP-SMA (10/15, 66.7%). An earlier disease onset and a family history of the disease or consanguinity were independent predictors of a positive genetic diagnosis. Neuropathy gene panels were performed in 59 patients with a 32.2% diagnostic yield (19/59). In 13 additional patients, a genetic diagnosis was achieved through individual gene sequencing or an alternative neuromuscular NGS.
Discussion: Non-5q SMA is genetically heterogeneous, and neuropathy gene panels achieve a molecular diagnosis in one-third of the patients. The diagnostic yield can be increased by sequencing of other neuromuscular and neurometabolic genes. Nevertheless, there is an unmet need to cluster these patients to aid in the identification of new genes.
Copyright © 2023 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Conflict of interest statement
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
Figures
References
-
- Pipis M, Rossor AM, Laura M, Reilly MM. Next-generation sequencing in Charcot–Marie–Tooth disease: opportunities and challenges. Nat Rev Neurol. 2019;15(11):644-656. - PubMed
-
- Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6-14. - PubMed
-
- Beijer D, Baets J. The expanding genetic landscape of hereditary motor neuropathies. Brain. 2020;143(12):3540-3563. - PubMed
-
- Brzustowicz LM, Lehner T, Castilla LH, et al. . Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q1 1.2-13.3. Nature. 1990;344(6266):540-541. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous