

A macroecological perspective on genetic diversity in the human gut microbiome
- PMID: 37478102
- PMCID: PMC10361512
- DOI: 10.1371/journal.pone.0288926
A macroecological perspective on genetic diversity in the human gut microbiome
Abstract
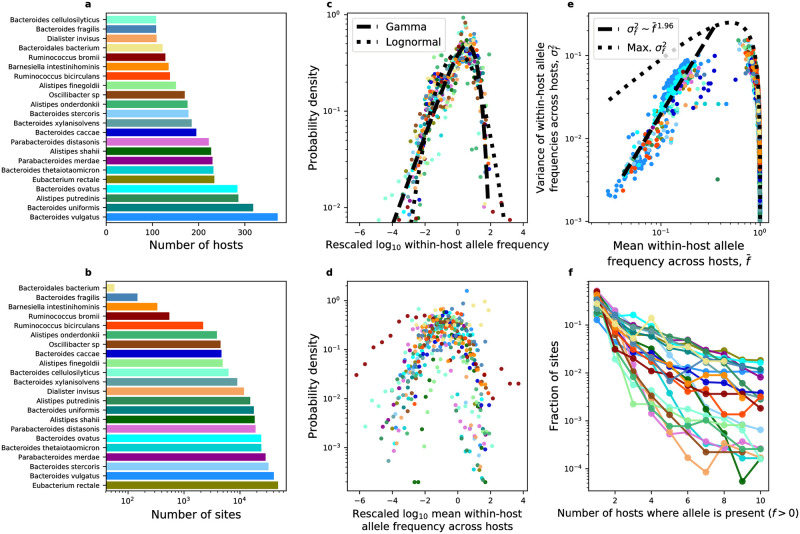
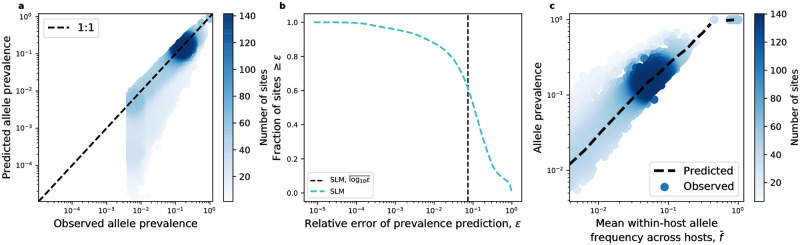
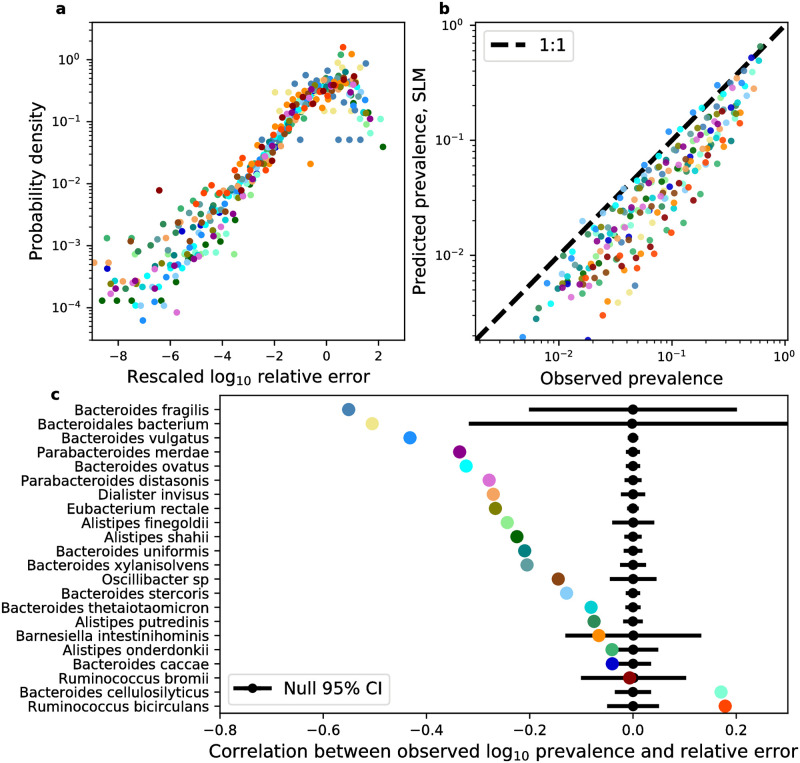
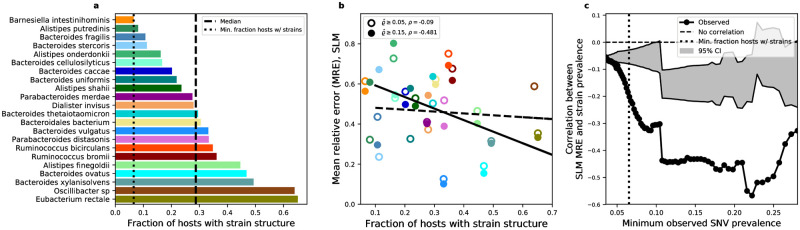
While the human gut microbiome has been intensely studied, we have yet to obtain a sufficient understanding of the genetic diversity that it harbors. Research efforts have demonstrated that a considerable fraction of within-host genetic variation in the human gut is driven by the ecological dynamics of co-occurring strains belonging to the same species, suggesting that an ecological lens may provide insight into empirical patterns of genetic diversity. Indeed, an ecological model of self-limiting growth and environmental noise known as the Stochastic Logistic Model (SLM) was recently shown to successfully predict the temporal dynamics of strains within a single human host. However, its ability to predict patterns of genetic diversity across human hosts has yet to be tested. In this manuscript I determine whether the predictions of the SLM explain patterns of genetic diversity across unrelated human hosts for 22 common microbial species. Specifically, the stationary distribution of the SLM explains the distribution of allele frequencies across hosts and predicts the fraction of hosts harboring a given allele (i.e., prevalence) for a considerable fraction of sites. The accuracy of the SLM was correlated with independent estimates of strain structure, suggesting that patterns of genetic diversity in the gut microbiome follow statistically similar forms across human hosts due to the existence of strain-level ecology.
Copyright: © 2023 William R. Shoemaker. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Ecological Stability Emerges at the Level of Strains in the Human Gut Microbiome.mBio. 2023 Apr 25;14(2):e0250222. doi: 10.1128/mbio.02502-22. Epub 2023 Feb 21. mBio. 2023. PMID: 36809109 Free PMC article.
-
Universal gut microbial relationships in the gut microbiome of wild baboons.Elife. 2023 May 9;12:e83152. doi: 10.7554/eLife.83152. Elife. 2023. PMID: 37158607 Free PMC article.
-
Interpopulation Variation in the Atlantic Salmon Microbiome Reflects Environmental and Genetic Diversity.Appl Environ Microbiol. 2018 Aug 1;84(16):e00691-18. doi: 10.1128/AEM.00691-18. Print 2018 Aug 15. Appl Environ Microbiol. 2018. PMID: 29915104 Free PMC article.
-
Population Genetics in the Human Microbiome.Trends Genet. 2020 Jan;36(1):53-67. doi: 10.1016/j.tig.2019.10.010. Epub 2019 Nov 25. Trends Genet. 2020. PMID: 31780057 Review.
-
Global landscape of gut microbiome diversity and antibiotic resistomes across vertebrates.Sci Total Environ. 2022 Sep 10;838(Pt 2):156178. doi: 10.1016/j.scitotenv.2022.156178. Epub 2022 May 23. Sci Total Environ. 2022. PMID: 35618126 Review.
Cited by
-
Investigating macroecological patterns in coarse-grained microbial communities using the stochastic logistic model of growth.Elife. 2024 Jan 22;12:RP89650. doi: 10.7554/eLife.89650. Elife. 2024. PMID: 38251984 Free PMC article.
-
Sparse species interactions reproduce abundance correlation patterns in microbial communities.Proc Natl Acad Sci U S A. 2024 Jan 30;121(5):e2309575121. doi: 10.1073/pnas.2309575121. Epub 2024 Jan 24. Proc Natl Acad Sci U S A. 2024. PMID: 38266051 Free PMC article.
-
Macroecological patterns in experimental microbial communities.PLoS Comput Biol. 2025 May 8;21(5):e1013044. doi: 10.1371/journal.pcbi.1013044. eCollection 2025 May. PLoS Comput Biol. 2025. PMID: 40341906 Free PMC article.
-
The characteristics of intestinal microflora in infants with rotavirus enteritis, changes in microflora before and after treatment and their clinical values.Sci Rep. 2025 Feb 5;15(1):4312. doi: 10.1038/s41598-025-88312-w. Sci Rep. 2025. PMID: 39910252 Free PMC article.
References
-
- Ghosh OM, Good BH. Emergent evolutionary forces in spatial models of luminal growth in the human gut microbiota; 2021. Available from: https://www.biorxiv.org/content/10.1101/2021.07.15.452569v1. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
