

Genome-scale mapping of DNA damage suppressors through phenotypic CRISPR-Cas9 screens
- PMID: 37478847
- PMCID: PMC10530064
- DOI: 10.1016/j.molcel.2023.06.025
Genome-scale mapping of DNA damage suppressors through phenotypic CRISPR-Cas9 screens
Abstract
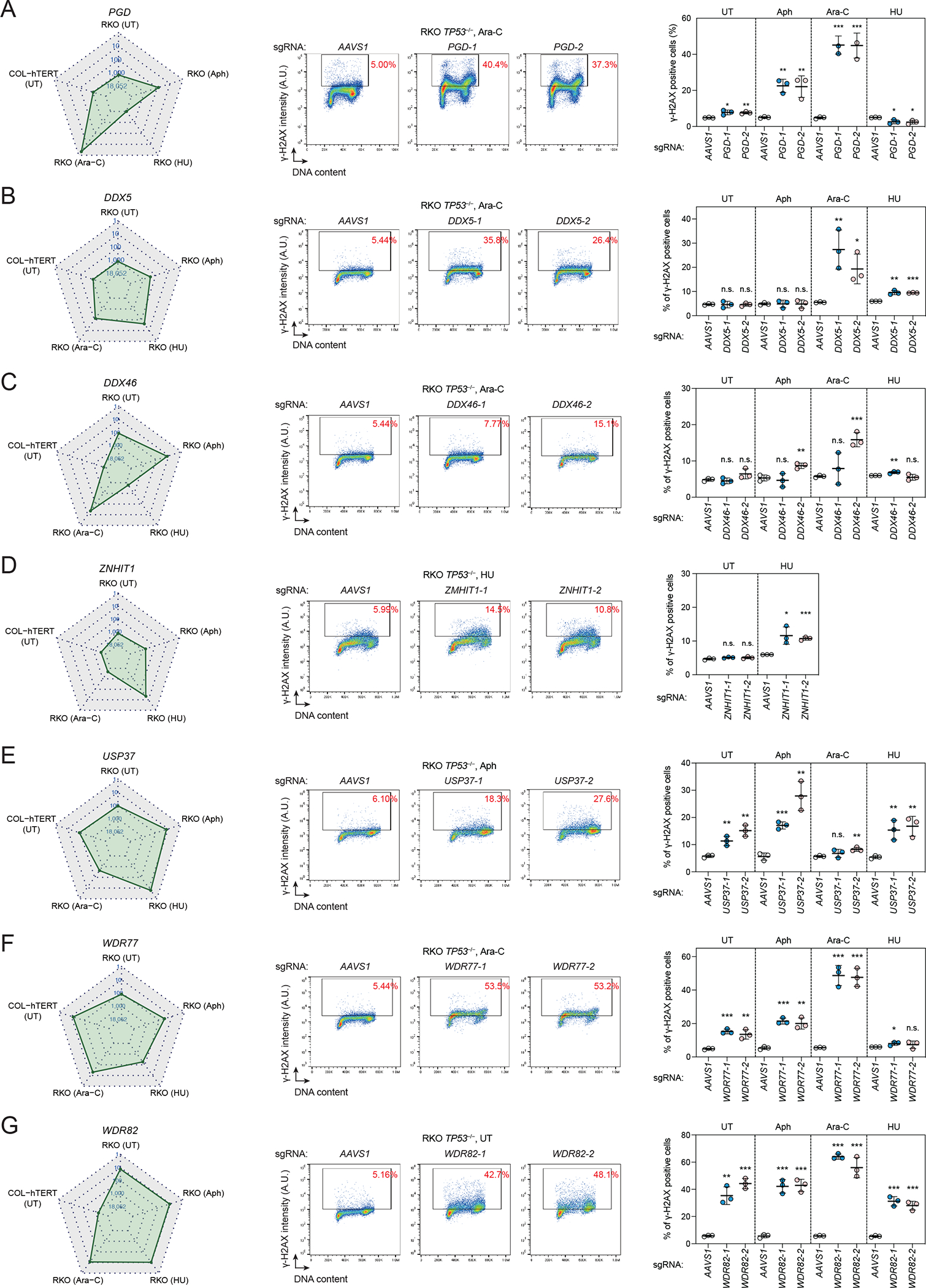
To maintain genome integrity, cells must accurately duplicate their genome and repair DNA lesions when they occur. To uncover genes that suppress DNA damage in human cells, we undertook flow-cytometry-based CRISPR-Cas9 screens that monitored DNA damage. We identified 160 genes whose mutation caused spontaneous DNA damage, a list enriched in essential genes, highlighting the importance of genomic integrity for cellular fitness. We also identified 227 genes whose mutation caused DNA damage in replication-perturbed cells. Among the genes characterized, we discovered that deoxyribose-phosphate aldolase DERA suppresses DNA damage caused by cytarabine (Ara-C) and that GNB1L, a gene implicated in 22q11.2 syndrome, promotes biogenesis of ATR and related phosphatidylinositol 3-kinase-related kinases (PIKKs). These results implicate defective PIKK biogenesis as a cause of some phenotypes associated with 22q11.2 syndrome. The phenotypic mapping of genes that suppress DNA damage therefore provides a rich resource to probe the cellular pathways that influence genome maintenance.
Keywords: ATR; DNA damage; DNA replication stress; TTT complex; cytarabine; genome stability.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.D. is a shareholder and advisor for Repare Therapeutics. A.-C.G. is a member of Molecular Cell’s Advisory Board.
Figures






References
-
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. - PubMed
-
- Ward IM, and Chen J (2001). Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276, 47759–47762. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
