

Screening for lysosomal diseases in a selected pediatric population: the case of Gaucher disease and acid sphingomyelinase deficiency
- PMID: 37480063
- PMCID: PMC10362631
- DOI: 10.1186/s13023-023-02797-0
Screening for lysosomal diseases in a selected pediatric population: the case of Gaucher disease and acid sphingomyelinase deficiency
Abstract
Background: GD and ASMD are lysosomal storage disorders that enter into differential diagnosis due to the possible overlap in their clinical manifestations. The availability of safe and effective enzymatic therapies has recently led many investigators to develop and validate new screening tools, such as algorithms, for the diagnosis of LSDs where the lack of disease awareness or failure to implement newborn screening results in a delayed diagnosis.
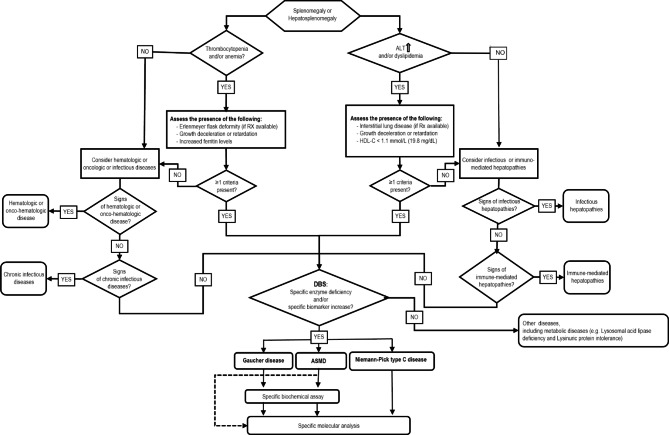
Results: the proposed algorithm allows for the clinical and biochemical differentiation between GD and ASMD. It is based on enzyme activity assessed on dried blood spots by multiplexed tandem mass spectrometry (MS/MS) coupled to specific biomarkers as second-tier analysis.
Conclusions: we believe that this method will provide a simple, convenient and sensitive tool for the screening of a selected population that can be used by pediatricians and other specialists (such as pediatric hematologists and pediatric hepatologists) often engaged in diagnosing these disorders.
Keywords: Acid Sphingomyelinase Deficiency; Gaucher Disease; Lysosomal Storage Diseases; Selected population screening; Splenomegaly.
© 2023. The Author(s).
Figures
References
-
- Pastores G, Hughes D. Gaucher Disease - GeneReviews® - NCBI Bookshelf [Internet]. GeneReviews. 2018 [cited 2022 Sep 27]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1269/.
-
- Kaplan P, Baris H, De Meirleir L, Di Rocco M, El-Beshlawy A, Huemer M et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr [Internet]. Eur J Pediatr; 2013 [cited 2022 Sep 27];172:447–58. Available from: https://pubmed.ncbi.nlm.nih.gov/22772880/. - PubMed
-
- Wasserstein M, Dionisi-Vici C, Giugliani R, Hwu WL, Lidove O, Lukacs Z et al. Recommendations for clinical monitoring of patients with acid sphingomyelinase deficiency (ASMD). Mol Genet Metab [Internet]. Mol Genet Metab; 2019 [cited 2022 Sep 27];126:98–105. Available from: https://pubmed.ncbi.nlm.nih.gov/30514648/. - PMC - PubMed
-
- McGovern MM, Avetisyan R, Sanson BJ, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis [Internet]. BioMed Central Ltd.; 2017 [cited 2022 Sep 27];12:1–13. Available from: https://ojrd.biomedcentral.com/articles/10.1186/s13023-017-0572-x. - PMC - PubMed
-
- Wasserstein MP, Diaz GA, Lachmann RH, Jouvin MH, Nandy I, Ji AJ et al. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): safety and efficacy in adults treated for 30 months. J Inherit Metab Dis [Internet]. J Inherit Metab Dis; 2018 [cited 2022 Sep 27];41:829–38. Available from: https://pubmed.ncbi.nlm.nih.gov/29305734/. - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
