

A Comparative Analysis of Data Analysis Tools for Data-Independent Acquisition Mass Spectrometry
- PMID: 37481071
- PMCID: PMC10458344
- DOI: 10.1016/j.mcpro.2023.100623
A Comparative Analysis of Data Analysis Tools for Data-Independent Acquisition Mass Spectrometry
Abstract
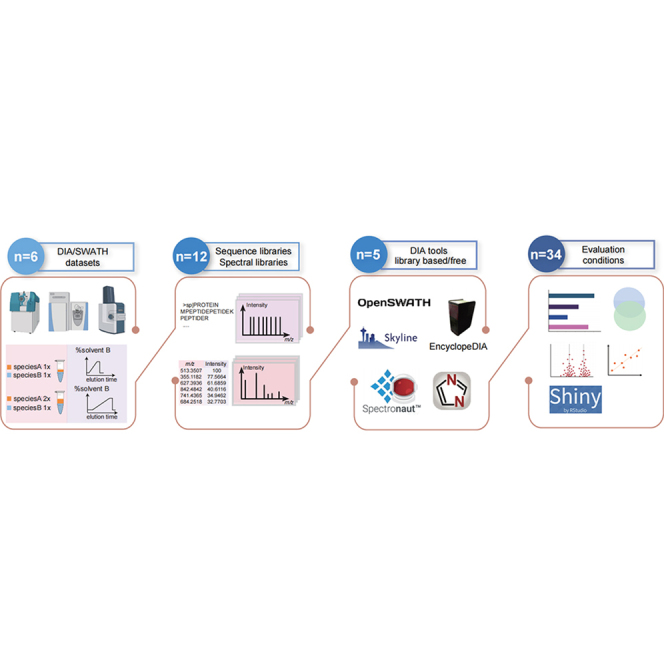
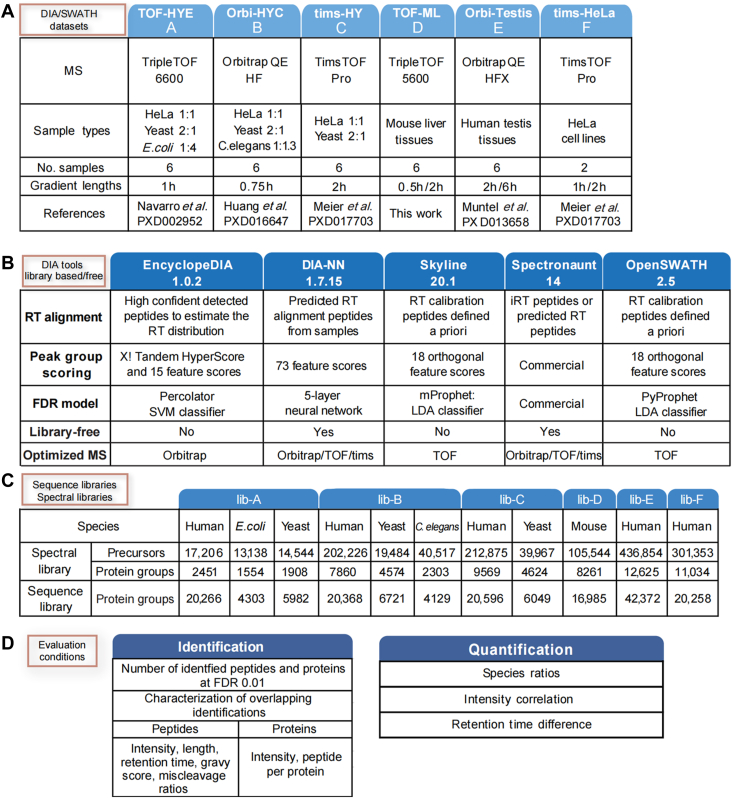
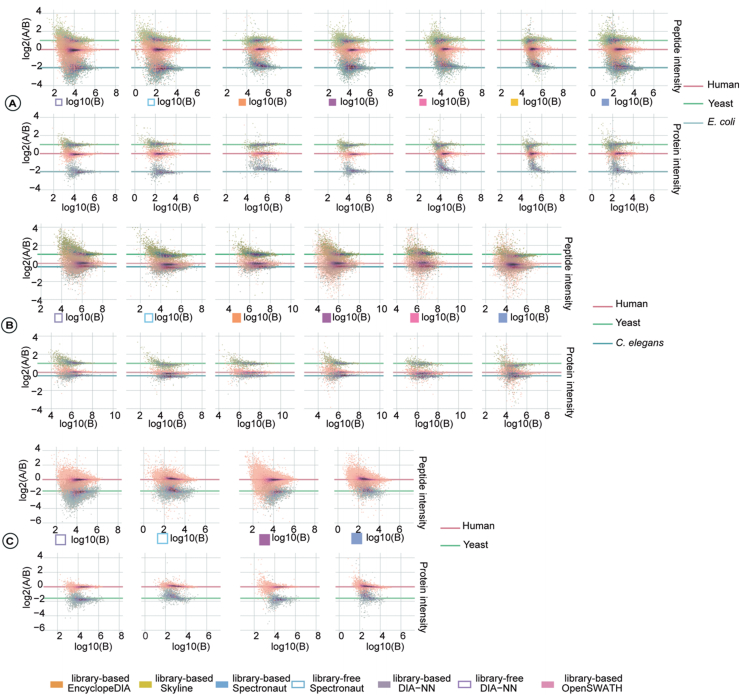
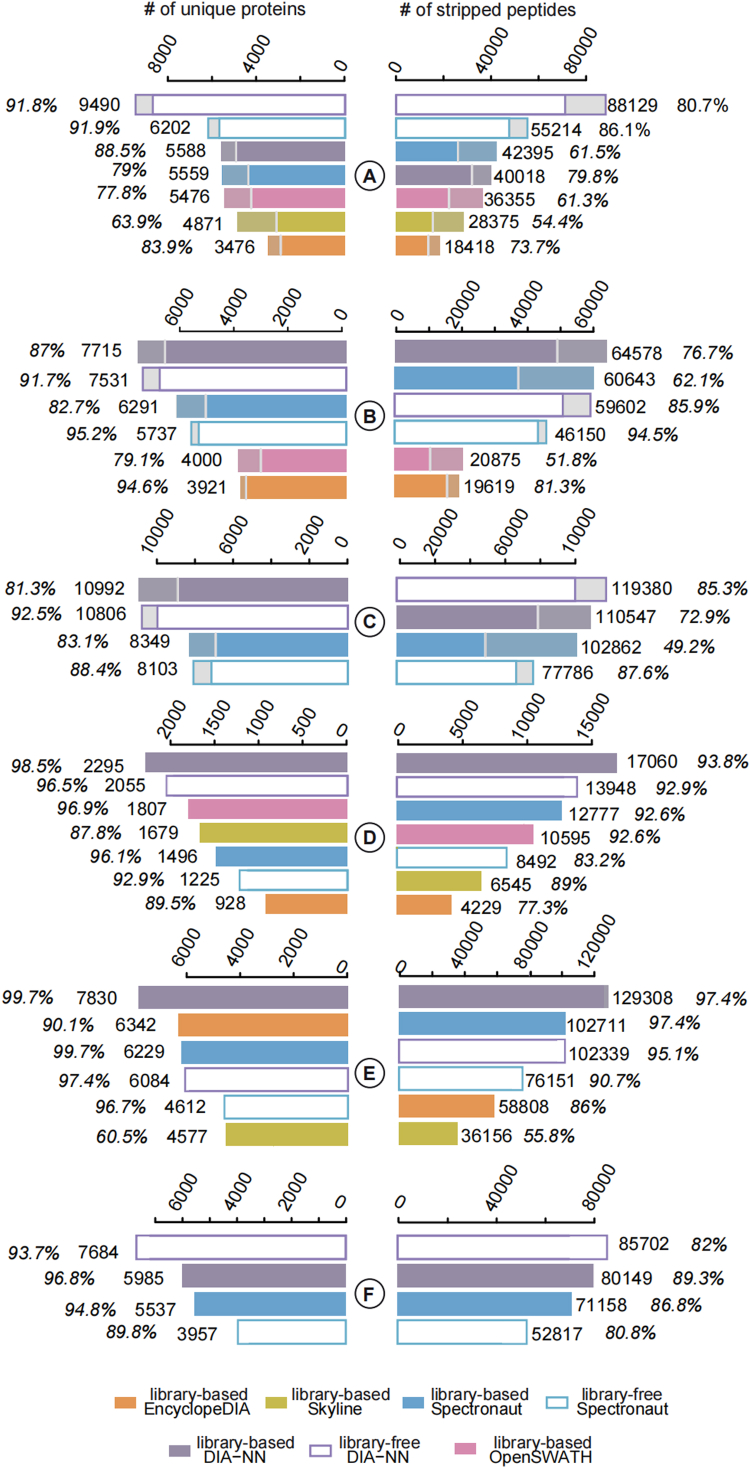
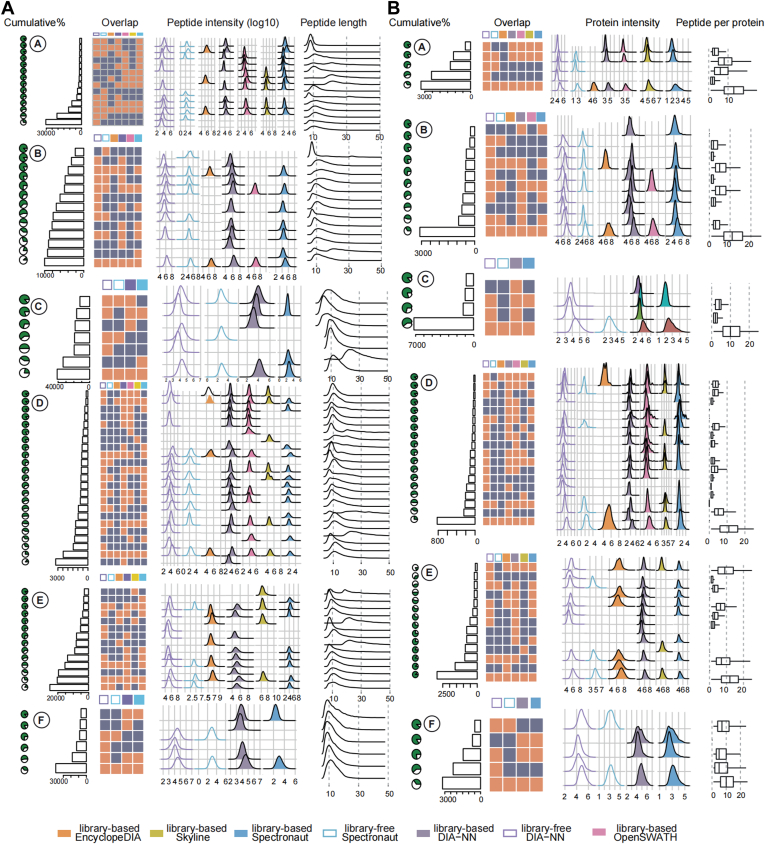
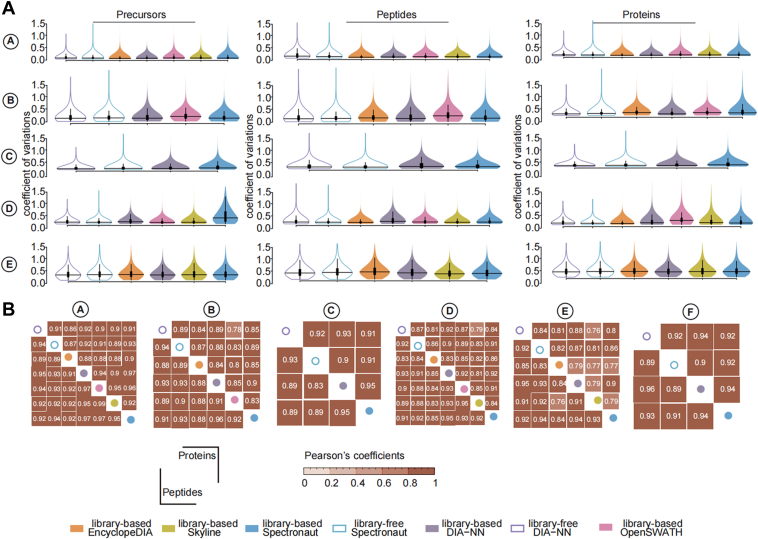
Data-independent acquisition (DIA) mass spectrometry-based proteomics generates reproducible proteome data. The complex processing of the DIA data has led to the development of multiple data analysis tools. In this study, we assessed the performance of five tools (OpenSWATH, EncyclopeDIA, Skyline, DIA-NN, and Spectronaut) using six DIA datasets obtained from TripleTOF, Orbitrap, and TimsTOF Pro instruments. By comparing identification and quantification metrics and examining shared and unique cross-tool identifications, we evaluated both library-based and library-free approaches. Our findings indicate that library-free approaches outperformed library-based methods when the spectral library had limited comprehensiveness. However, our results also suggest that constructing a comprehensive library still offers benefits for most DIA analyses. This study provides comprehensive guidance for DIA data analysis tools, benefiting both experienced and novice users of DIA-mass spectrometry technology.
Keywords: DIA-NN; EncyclopeDIA; OpenSWATH; Skyline; Spectronaut; data-independent acquisition; mass spectrometry; proteomics.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest T. G. is shareholder of Westlake Omics, Inc. W. G., L. H., D. L., and L. L. were employees of Westlake Omics, Inc. when they participated in this project.
Figures
References
-
- Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. - PubMed
-
- Meier F., Brunner A.D., Frank M., Ha A., Bludau I., Voytik E., et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. - PubMed
-
- Venable J.D., Dong M.Q., Wohlschlegel J., Dillin A., Yates J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods. 2004;1:39–45. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
