

Should evidence of an autolysosomal de-acidification defect in Alzheimer and Parkinson diseases call for caution in prescribing chronic PPI and DMARD?
- PMID: 37482676
- PMCID: PMC10472882
- DOI: 10.1080/15548627.2023.2214960
Should evidence of an autolysosomal de-acidification defect in Alzheimer and Parkinson diseases call for caution in prescribing chronic PPI and DMARD?
Abstract
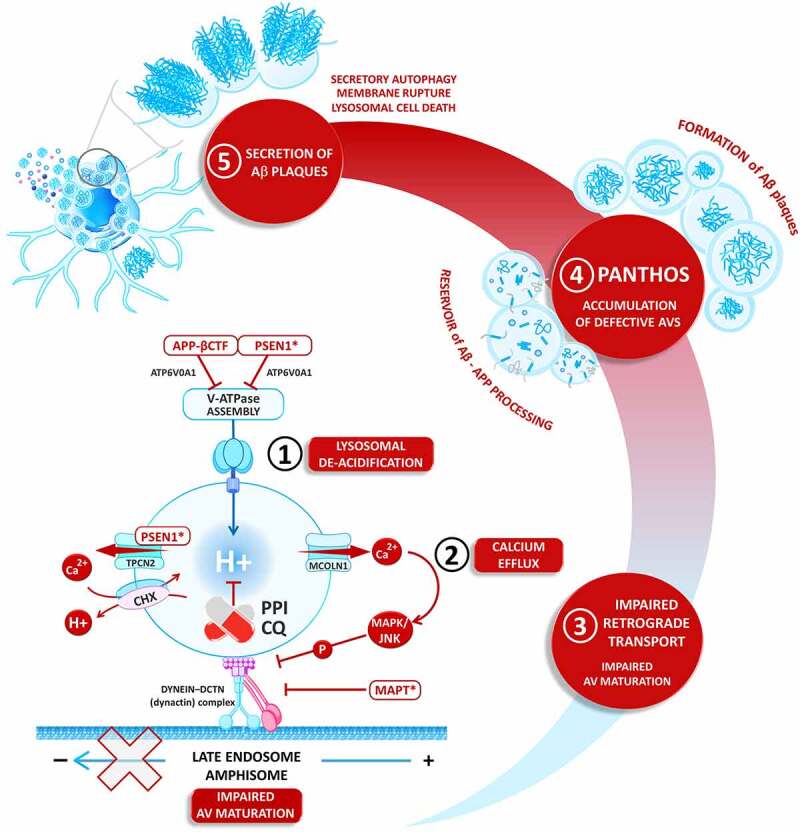
Nearly fifty million older people suffer from neurodegenerative diseases, including Alzheimer (AD) and Parkinson (PD) disease, a global burden expected to triple by 2050. Such an imminent "neurological pandemic" urges the identification of environmental risk factors that are hopefully avoided to fight the disease. In 2022, strong evidence in mouse models incriminated defective lysosomal acidification and impairment of the autophagy pathway as modifiable risk factors for dementia. To date, the most prescribed lysosomotropic drugs are proton pump inhibitors (PPIs), chloroquine (CQ), and the related hydroxychloroquine (HCQ), which belong to the group of disease-modifying antirheumatic drugs (DMARDs). This commentary aims to open the discussion on the possible mechanisms connecting the long-term prescribing of these drugs to the elderly and the incidence of neurodegenerative diseases.Abbreviations: AD: Alzheimer disease; APP-βCTF: amyloid beta precursor protein-C-terminal fragment; BACE1: beta-secretase 1; BBB: brain blood barrier; CHX: Ca2+/H+ exchanger; CMI: cognitive mild impairment; CQ: chloroquine; DMARD: disease-modifying antirheumatic drugs; GBA1: glucosylceramidase beta 1; HCQ: hydroxychloroquine; HPLC: high-performance liquid chromatography; LAMP: lysosomal associated membrane protein; MAPK/JNK: mitogen-activated protein kinase; MAPT: microtubule associated protein tau; MCOLN1/TRPML1: mucolipin TRP cation channel 1; NFE2L2/NRF2: NFE2 like bZIP transcription factor 2; NRBF2: nuclear receptor binding factor 2; PANTHOS: poisonous flower; PD: Parkinson disease; PIK3C3: phosphatIdylinositol 3-kinase catalytic subunit type 3; PPI: proton pump inhibitor; PSEN1: presenilin 1, RUBCN: rubicon autophagy regulator; RUBCNL: rubicon like autophagy enhancer; SQSTM1: sequestosome 1; TMEM175: transmembrane protein 175; TPCN2: two pore segment channel 2; VATPase: vacuolar-type H+-translocating ATPase; VPS13C: vacuolar protein sorting ortholog 13 homolog C; VPS35: VPS35 retromer complex component; WDFY3: WD repeat and FYVE domain containing 3; ZFYVE1: zinc finger FYVE-type containing 1.
Keywords: Autophagy; disease-modifying antirheumatic drugs; lysosome; neurodegenerative disease; proton pump inhibitor.
Conflict of interest statement
No potential conflict of interest was reported by the authors.
Figures
References
-
- Kong X, Chen L, Jiao L, et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J Med Chem. 2014;57:9512–9521. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous