

Inaugurating High-Throughput Profiling of Extracellular Vesicles for Earlier Ovarian Cancer Detection
- PMID: 37485618
- PMCID: PMC10520636
- DOI: 10.1002/advs.202301930
Inaugurating High-Throughput Profiling of Extracellular Vesicles for Earlier Ovarian Cancer Detection
Abstract
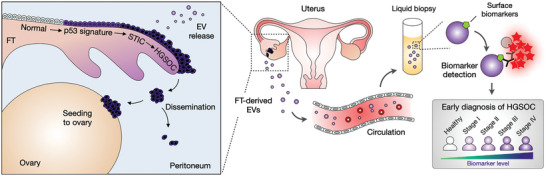
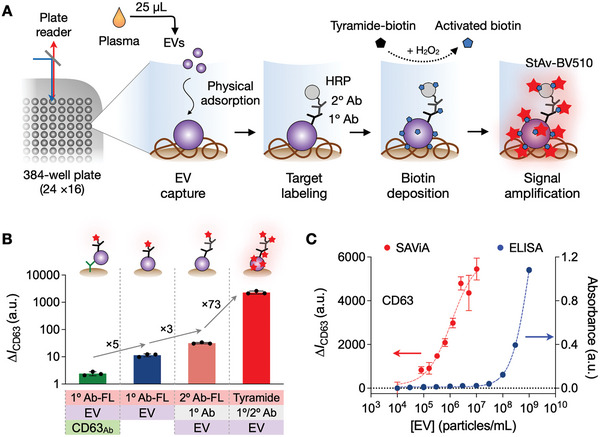
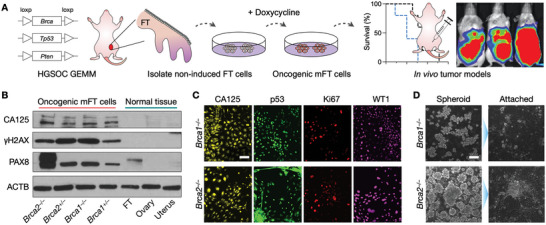
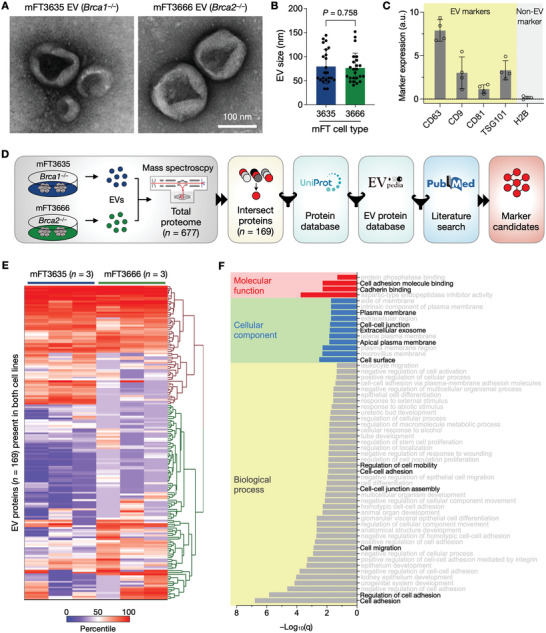
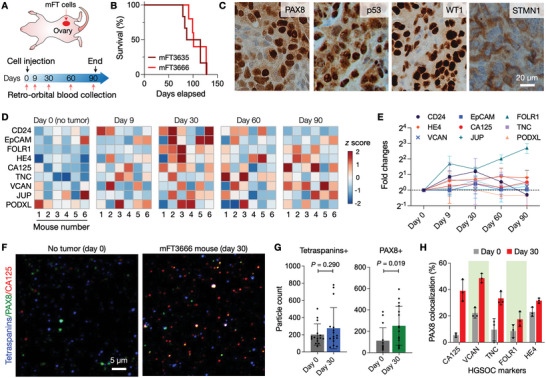
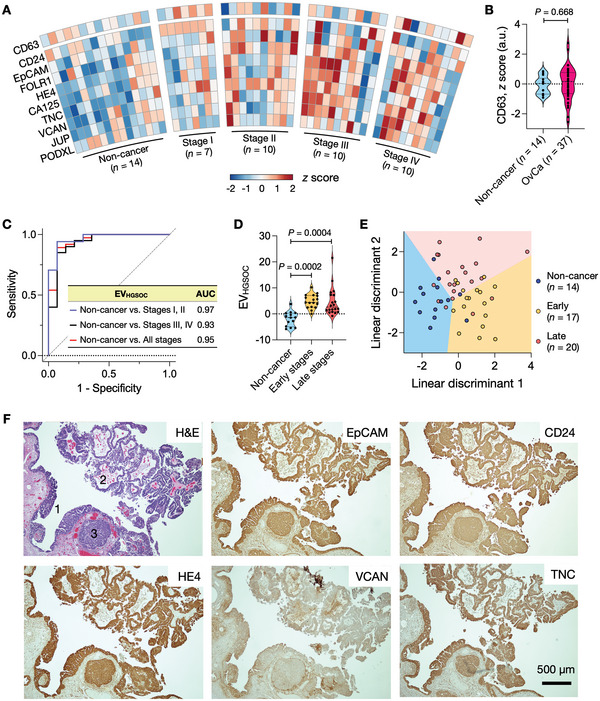
Detecting early cancer through liquid biopsy is challenging due to the lack of specific biomarkers for early lesions and potentially low levels of these markers. The current study systematically develops an extracellular-vesicle (EV)-based test for early detection, specifically focusing on high-grade serous ovarian carcinoma (HGSOC). The marker selection is based on emerging insights into HGSOC pathogenesis, notably that it arises from precursor lesions within the fallopian tube. This work thus establishes murine fallopian tube (mFT) cells with oncogenic mutations and performs proteomic analyses on mFT-derived EVs. The identified markers are then evaluated with an orthotopic HGSOC animal model. In serially-drawn blood of tumor-bearing mice, mFT-EV markers increase with tumor initiation, supporting their potential use in early cancer detection. A pilot clinical study (n = 51) further narrows EV markers to five candidates, EpCAM, CD24, VCAN, HE4, and TNC. The combined expression of these markers distinguishes HGSOC from non-cancer with 89% sensitivity and 93% specificity. The same markers are also effective in classifying three groups (non-cancer, early-stage HGSOC, and late-stage HGSOC). The developed approach, for the first time inaugurated in fallopian tube-derived EVs, could be a minimally invasive tool to monitor women at high risk of ovarian cancer for timely intervention.
Keywords: diagnostics; extracellular vesicles; ovarian cancer.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Menon U., Gentry‐Maharaj A., Burnell M., Singh N., Ryan A., Karpinskyj C., Carlino G., Taylor J., Massingham S. K., Raikou M., Kalsi J. K., Woolas R., Manchanda R., Arora R., Casey L., Dawnay A., Dobbs S., Leeson S., Mould T., Seif M. W., Sharma A., Williamson K., Liu Y., Fallowfield L., Mcguire A. J., Campbell S., Skates S. J., Jacobs I. J., Parmar M., Lancet 2021, 397, 2182. - PMC - PubMed
-
- Buys S. S., Patridge E., Black A., Johnson C. C., Lamerato L., Isaacs C., Reding D. J., Greenlee R. T., Yokochi L. A., Kessel B., Crawford E. D., Church T. R., Andriole G. L., Weissfeld J. L, Fouad M. N., Chia D., O'Brien B., Ragard L. R., Clapp J. D., Rathmell J. M., Riley T. L., Hartge P., Pinsky P. F., Zhu C. S., Izmirlian G., Kramer B. S., Miller A. B., Xu J.‐L., Prorok P. C., Gohagan J. K., et al., JAMA, J. Am. Med. Assoc. 2011, 305, 2295.
-
- Henderson J. T., Webber E. M., Sawaya G. F., JAMA, J. Am. Med. Assoc. 2018, 319, 595. - PubMed
-
- Labidi‐Galy S. I., Papp E., Hallberg D., Niknafs N., Adleff V., Noe M., Bhattacharya R., Novak M., Jones S., Phallen J., Hruban C. A., Hirsch M. S., Lin D. I., Schwartz L., Marie C. L., Tille J.‐C., Bowden M., Ayhan A., Wood L. D., Scharpf R. B., Kurman R., Wang T.‐L., Shih le‐M., Karchin R., Drapkin R., Velculescu V. E., Nat. Commun. 2017, 8, 1093. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous