

Blood cell traits' GWAS loci colocalization with variation in PU.1 genomic occupancy prioritizes causal noncoding regulatory variants
- PMID: 37492098
- PMCID: PMC10363807
- DOI: 10.1016/j.xgen.2023.100327
Blood cell traits' GWAS loci colocalization with variation in PU.1 genomic occupancy prioritizes causal noncoding regulatory variants
Abstract
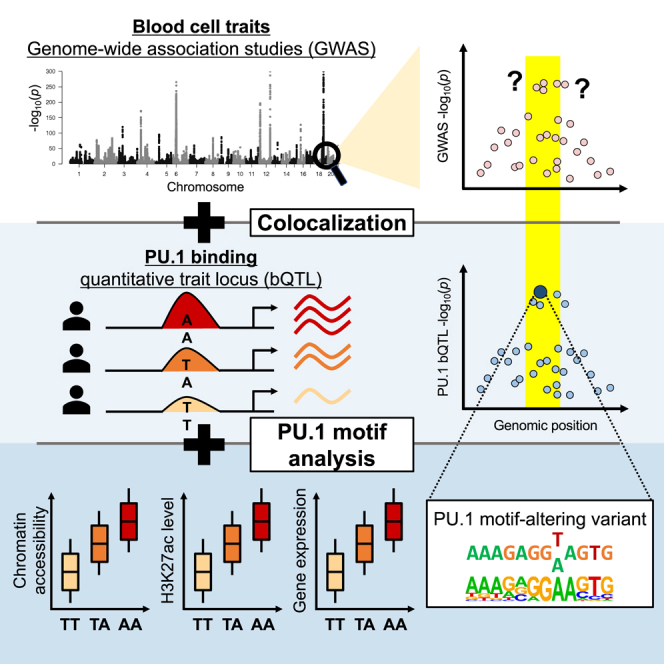
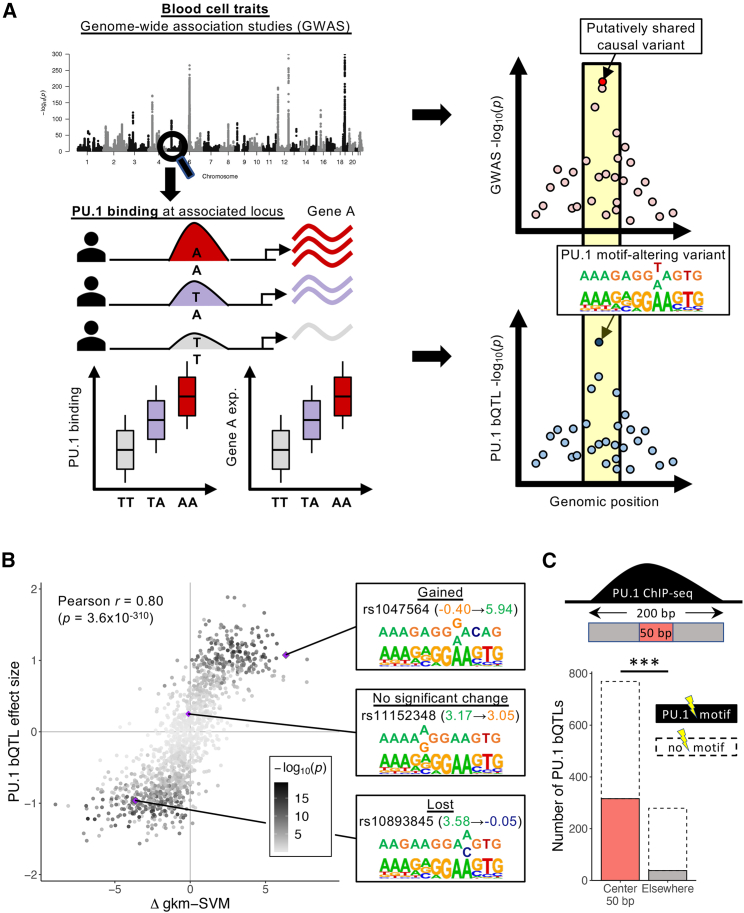
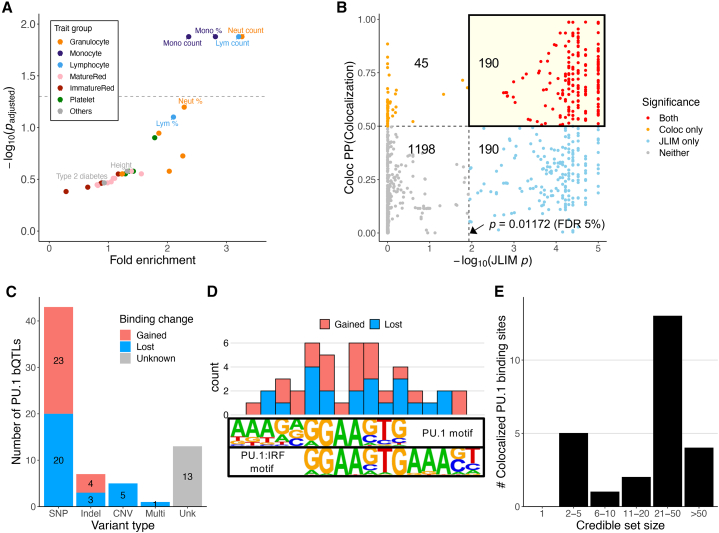
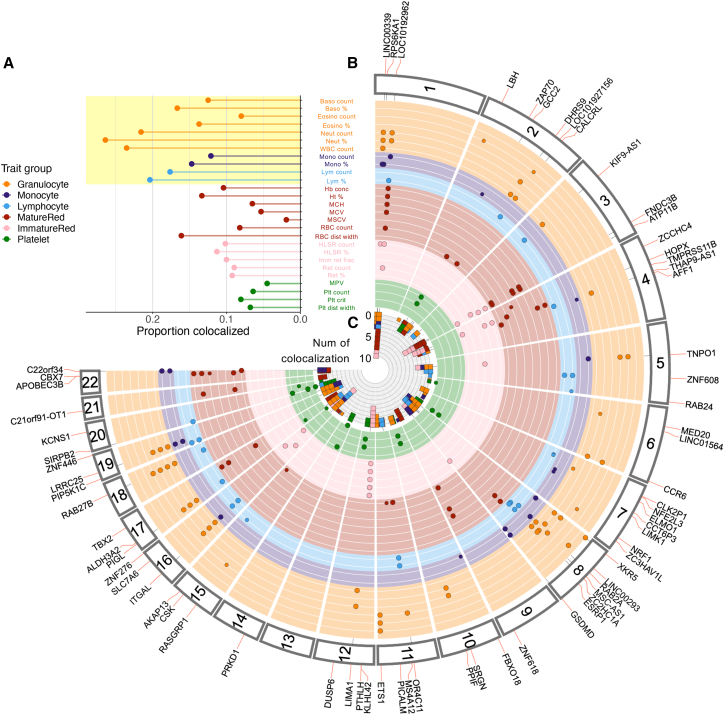
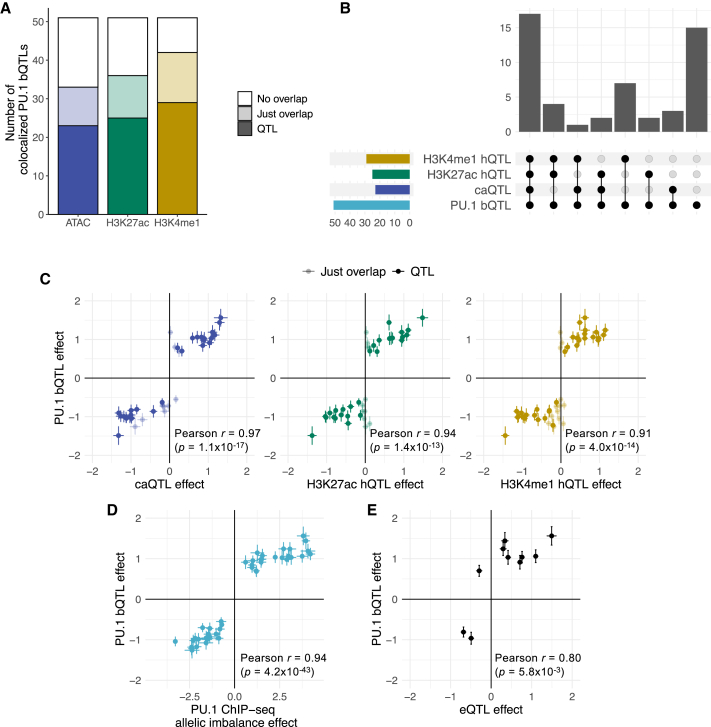
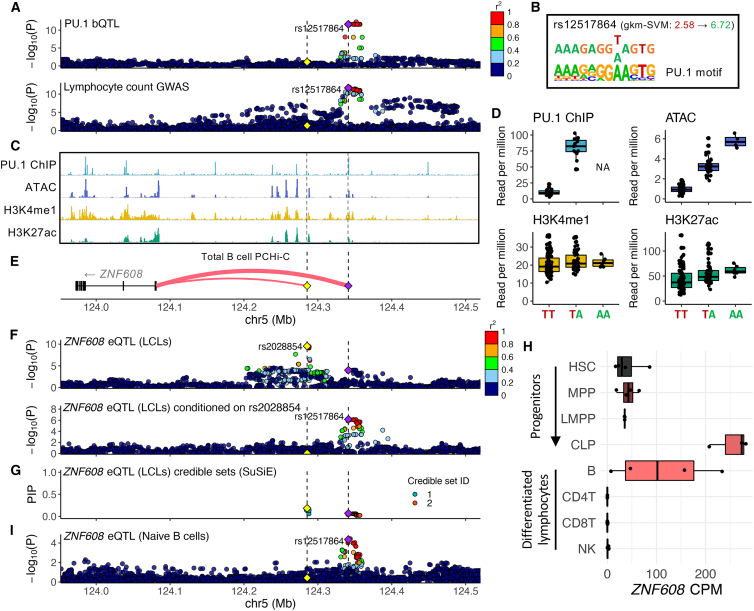
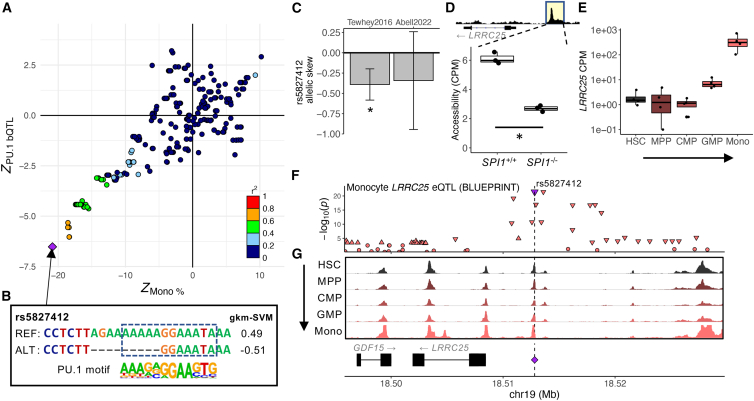
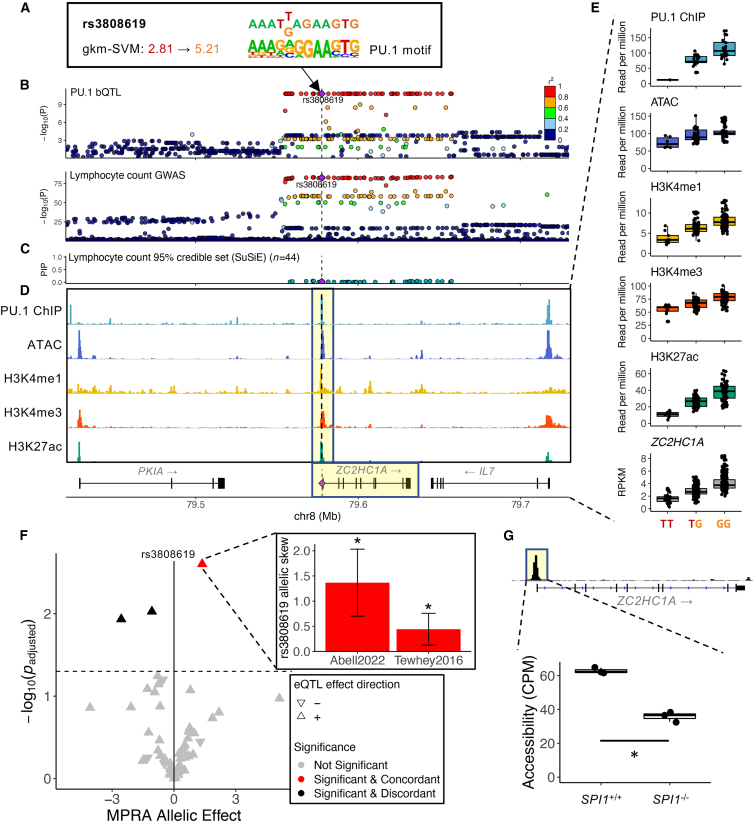
Genome-wide association studies (GWASs) have uncovered numerous trait-associated loci across the human genome, most of which are located in noncoding regions, making interpretation difficult. Moreover, causal variants are hard to statistically fine-map at many loci because of widespread linkage disequilibrium. To address this challenge, we present a strategy utilizing transcription factor (TF) binding quantitative trait loci (bQTLs) for colocalization analysis to identify trait associations likely mediated by TF occupancy variation and to pinpoint likely causal variants using motif scores. We applied this approach to PU.1 bQTLs in lymphoblastoid cell lines and blood cell trait GWAS data. Colocalization analysis revealed 69 blood cell trait GWAS loci putatively driven by PU.1 occupancy variation. We nominate PU.1 motif-altering variants as the likely shared causal variants at 51 loci. Such integration of TF bQTL data with other GWAS data may reveal transcriptional regulatory mechanisms and causal noncoding variants underlying additional complex traits.
Keywords: ChIP-seq; PU.1; TF bQTL; blood cell traits; colocalization; fine-mapping; genome-wide association study; noncoding variants; transcription factor binding quantitative trait locus; transcription factors; variant-to-function.
© 2023 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Colocalization of blood cell traits GWAS associations and variation in PU.1 genomic occupancy prioritizes causal noncoding regulatory variants.bioRxiv [Preprint]. 2023 Mar 30:2023.03.29.534582. doi: 10.1101/2023.03.29.534582. bioRxiv. 2023. Update in: Cell Genom. 2023 May 17;3(7):100327. doi: 10.1016/j.xgen.2023.100327. PMID: 37034747 Free PMC article. Updated. Preprint.
References
-
- International Common Disease Alliance International common disease alliance white paper v1.0. 2020. https://www.icda.bio
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
