

Immune escape and resistance to immunotherapy in mismatch repair deficient tumors
- PMID: 37492581
- PMCID: PMC10363668
- DOI: 10.3389/fimmu.2023.1210164
Immune escape and resistance to immunotherapy in mismatch repair deficient tumors
Abstract
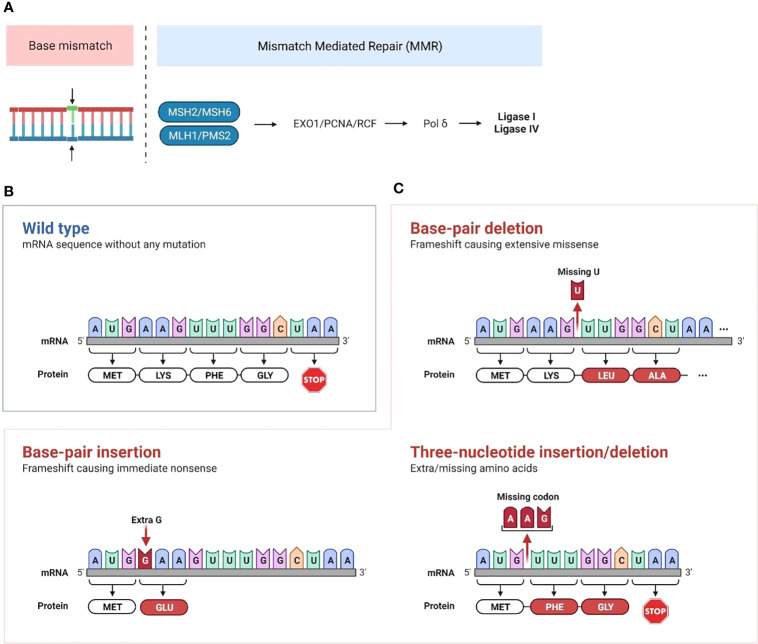
Up to 30% of colorectal, endometrial and gastric cancers have a deficiency in mismatch repair (MMR) protein expression due to either germline or epigenetic inactivation. Patients with Lynch Syndrome who inherit an inactive MMR allele have an up to 80% risk for developing a mismatch repair deficient (MMRd) cancer. Due to an inability to repair DNA, MMRd tumors present with genomic instability in microsatellite regions (MS). Tumors with high MS instability (MSI-H) are characterized by an increased frequency of insertion/deletions (indels) that can encode novel neoantigens if they occur in coding regions. The high tumor antigen burden for MMRd cancers is accompanied by an inflamed tumor microenvironment (TME) that contributes to the clinical effectiveness of anti-PD-1 therapy in this patient population. However, between 40 and 70% of MMRd cancer patients do not respond to treatment with PD-1 blockade, suggesting that tumor-intrinsic and -extrinsic resistance mechanisms may affect the success of checkpoint blockade. Immune evasion mechanisms that occur during early tumorigenesis and persist through cancer development may provide a window into resistance pathways that limit the effectiveness of anti-PD-1 therapy. Here, we review the mechanisms of immune escape in MMRd tumors during development and checkpoint blockade treatment, including T cell dysregulation and myeloid cell-mediated immunosuppression in the TME. Finally, we discuss the development of new therapeutic approaches to tackle resistance in MMRd tumors, including cancer vaccines, therapies targeting immunosuppressive myeloid programs, and immune checkpoint combination strategies.
Keywords: MMRd; MSI-H; PD1 (programmed cell death protein 1); colorectal cancer; immunotherapy; microsatellite unstable (high); myeloid cells; resistance.
Copyright © 2023 Mestrallet, Brown, Bozkus and Bhardwaj.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials